
Metagenomic Analysis of Wild Apple (Malus sieversii) Trees from Natural Habitats of Kazakhstan

 , ,
, ,
Abstract
1. Introduction
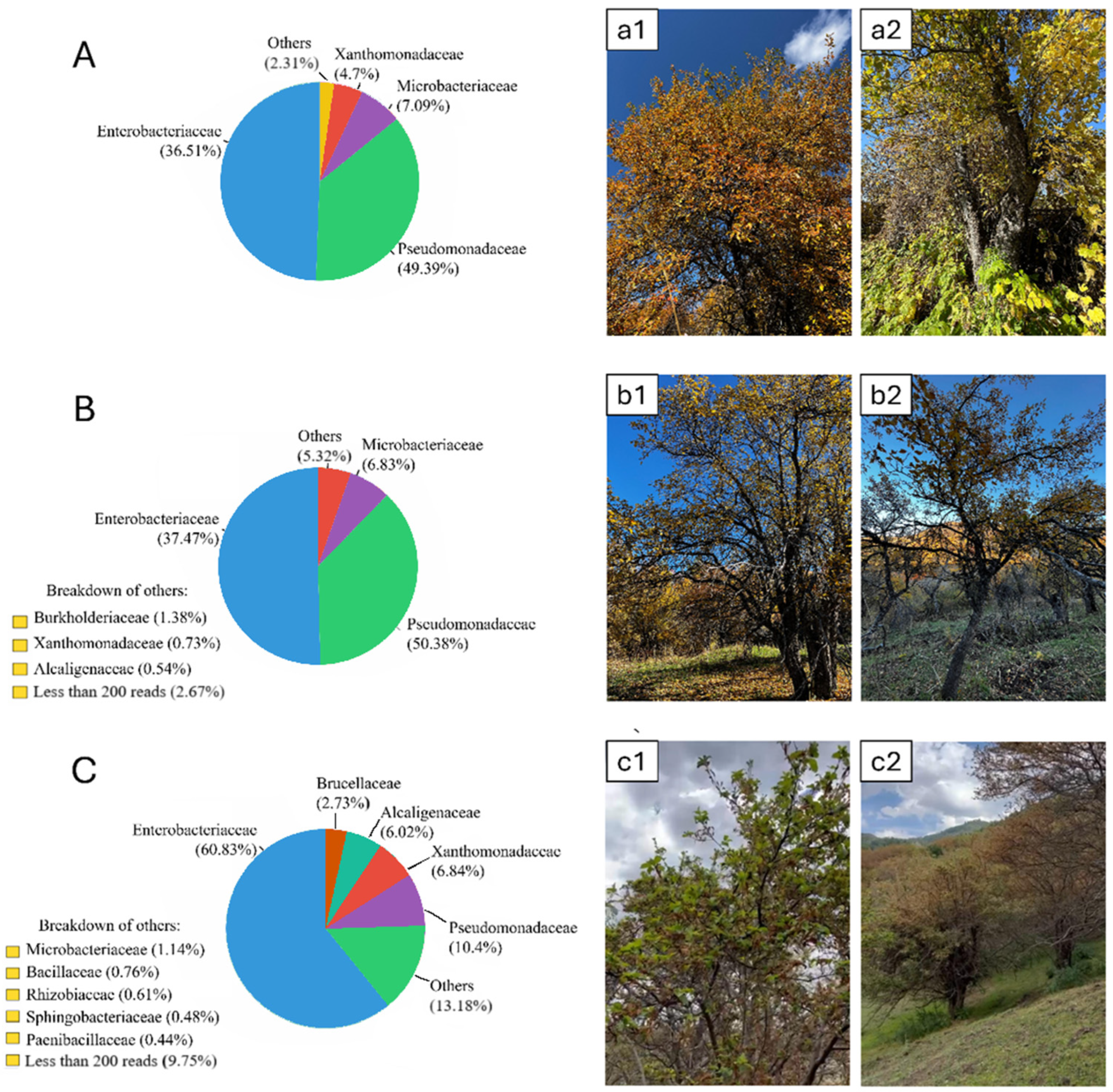
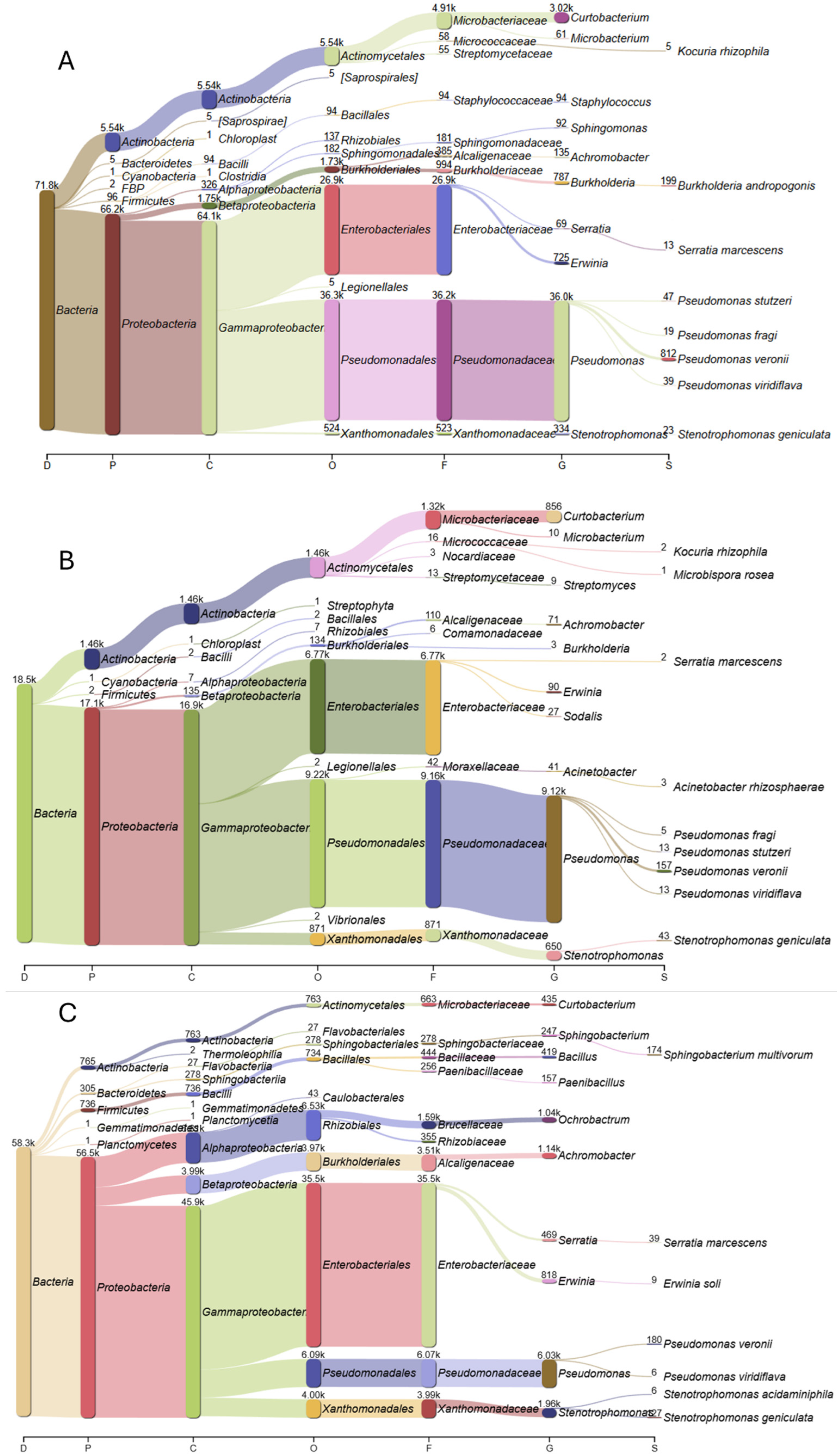
2. Results
3. Discussion
4. Materials and Methods
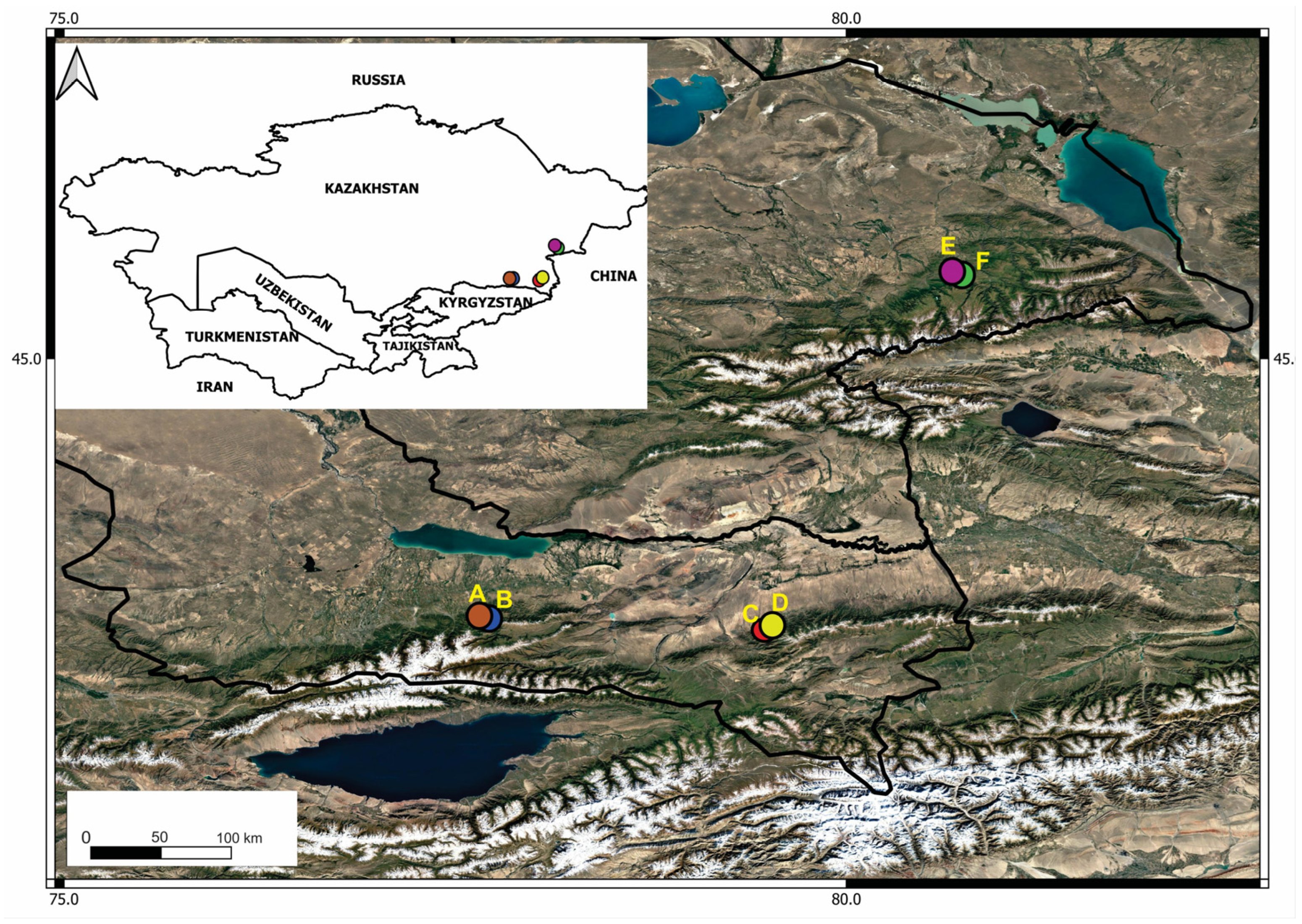
4.1. Collection Sites and Sampling
4.2. DNA Extraction
4.3. The Processes of 16S Amplification, Sequencing and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| IUCN | The International Union for Conservation of Nature’s |
| ONT | Oxford Nanopore Technology |
| PGPR | Plant growth-promoting rhizobacterium |
References
- Dzhangaliev, A.D. The Wild Apple Tree of Kazakhstan. In Horticultural Reviews; Janick, J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 29, pp. 63–303. ISBN 978-0-471-46337-5. [Google Scholar]
- Forsline, P.L.; Aldwinckle, H.S.; Dickson, E.E.; Luby, J.J.; Hokanson, S.C. Collection, Maintenance, Characterization, and Utilization of Wild Apples of Central Asia. In Horticultural Reviews; Janick, J., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2002; Volume 29, pp. 1–61. ISBN 978-0-471-46337-5. [Google Scholar]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The Genome of the Domesticated Apple (Malus × Domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Juniper, B.E.; Watkins, R.; Harris, S.A. The Origin of the Apple. Eucarpia Symp. Fruit Breed. Genet. 1998, 484, 27–33. [Google Scholar] [CrossRef]
- Duan, N.; Bai, Y.; Sun, H.; Wang, N.; Ma, Y.; Li, M.; Wang, X.; Jiao, C.; Legall, N.; Mao, L.; et al. Genome Re-Sequencing Reveals the History of Apple and Supports a Two-Stage Model for Fruit Enlargement. Nat. Commun. 2017, 8, 249. [Google Scholar] [CrossRef]
- Volume 2: Plants. In Red Book of Kazakhstan, 2nd ed.; Revised and Supplemented; Publishing House: Astana, Kazakhstan, 2014.
- IUCN. Malus sieversii. The IUCN Red List of Threatened Species. 2007, e.T32363A9693009. Available online: https://www.iucnredlist.org/species/32363/9693009 (accessed on 15 March 2025).
- Eastwood, A.; Lazkov, G.; Newton, A.C. The Red List of Trees of Central Asia; Fauna and Flora International: Cambridge, UK; Botanic Gardens Conservation International: Kew, UK; IUCN Species Survival Commission (SSC): Hertfordshire Zoo, UK; Global Tree Specialist Group: Cambridge, UK; Global Tree Campaign: Chatteris, UK, 2009. [Google Scholar]
- Jarosz, A.M.; Davelos, A.L. Effects of disease in wild plant populations and the evolution of pathogen aggressiveness. New Phytol. 1995, 129, 371–387. [Google Scholar] [CrossRef]
- Harshman, J.M.; Evans, K.M.; Allen, H.; Potts, R.; Flamenco, J.; Aldwinckle, H.S.; Wisniewski, M.E.; Norelli, J.L. Fire Blight Resistance in Wild Accessions of Malus sieversii. Plant Dis. 2017, 101, 1738–1745. [Google Scholar] [CrossRef]
- Jiao, H.; Liu, L.; Wang, R.; Qin, W.; Zhang, B. The Rhizosphere Microbiome of Malus sieversii (Ldb.) Roem. in the Geographic and Environmental Gradients of China’s Xinjiang. BMC Microbiol. 2023, 23, 26. [Google Scholar] [CrossRef]
- Jiao, H.; Wang, R.; Qin, W.; Yang, J. Screening of Rhizosphere Nitrogen Fixing, Phosphorus and Potassium Solubilizing Bacteria of Malus sieversii (Ldb.) Roem. and the Effect on Apple Growth. J. Plant Physiol. 2024, 292, 154142. [Google Scholar] [CrossRef]
- Jiao, H.-Y.; Liu, L.; Yang, J.-X.; Qin, W.; Wang, R.-Z. Effects of Rhizosphere Nitrogen-Fixing, Phosphate-Solubilizing and Potassium-Solubilizing Bacteria on Leaf Nutrients and Physiological Traits in Different Natural Populations of Malus sieversii. Chin. J. Plant Ecol. 2023, 48, 930–942. [Google Scholar] [CrossRef]
- Zhang, H.; Xi, J.; Mahmoud Ali, H.S.; Zhao, F.; Yu, S.; Yu, K. Responses of Nutrients and Bacterial Communities to Temperature and Nitrogen Addition in Rhizosphere Soil for Malus sieversii Seedlings. J. Soil Sci. Plant Nutr. 2024, 24, 2786–2797. [Google Scholar] [CrossRef]
- Zhang, H.; Phillip, F.O.; Wu, L.; Zhao, F.; Yu, S.; Yu, K. Effects of Temperature and Nitrogen Application on Carbon and Nitrogen Accumulation and Bacterial Community Composition in Apple Rhizosphere Soil. Front. Plant Sci. 2022, 13, 859395. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Tack, A.J.M.; Wasserman, B.; Liu, J.; Berg, G.; Norelli, J.; Droby, S.; Wisniewski, M. Evidence for Host–Microbiome Co-Evolution in Apple. New Phytol. 2021, 234, 2088–2100. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Ma, S.; Cao, J.; Liao, H.; Huang, Q.; Chen, W. The Newest Oxford Nanopore R10.4.1 Full-Length 16S rRNA Sequencing Enables the Accurate Resolution of Species-Level Microbial Community Profiling. Appl. Environ. Microbiol. 2023, 89, e00605-23. [Google Scholar] [CrossRef]
- Hu, Y.; Green, G.S.; Milgate, A.W.; Stone, E.A.; Rathjen, J.P.; Schwessinger, B. Pathogen Detection and Microbiome Analysis of Infected Wheat Using a Portable DNA Sequencer. Phytobiomes J. 2019, 3, 92–101. [Google Scholar] [CrossRef]
- Nicotra, D.; Ghadamgahi, F.; Ghosh, S.; Anzalone, A.; Dimaria, G.; Mosca, A.; Massimino, M.E.; Vetukuri, R.R.; Catara, V. Genomic Insights and Biocontrol Potential of Ten Bacterial Strains from the Tomato Core Microbiome. Front. Plant Sci. 2024, 15, 1437947. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Liu, Y.; Zhou, X.; Yin, C.; Zhang, P.; Yang, Q.; Mao, L.; Shentu, X.; Yu, X. Nanopore Sequencing Technology and Its Application in Plant Virus Diagnostics. Front. Microbiol. 2022, 13, 939666. [Google Scholar] [CrossRef] [PubMed]
- Rolon, M.L.; Tan, X.; Chung, T.; Gonzalez-Escalona, N.; Chen, Y.; Macarisin, D.; LaBorde, L.F.; Kovac, J. The Composition of Environmental Microbiota in Three Tree Fruit Packing Facilities Changed over Seasons and Contained Taxa Indicative of L. monocytogenes Contamination. Microbiome 2023, 11, 128. [Google Scholar] [CrossRef]
- Pecman, A.; Adams, I.; Gutiérrez-Aguirre, I.; Fox, A.; Boonham, N.; Ravnikar, M.; Kutnjak, D. Systematic Comparison of Nanopore and Illumina Sequencing for the Detection of Plant Viruses and Viroids Using Total RNA Sequencing Approach. Front. Microbiol. 2022, 13, 883921. [Google Scholar] [CrossRef]
- Fanai, A.; Bohia, B.; Lalremruati, F.; Lalhriatpuii, N.; Lalrokimi; Lalmuanpuii, R.; Singh, P.K.; Zothanpuia. Plant growth promoting bacteria (PGPB)-induced plant adaptations to stresses: An updated review. PeerJ 2024, 20, 17882. [Google Scholar] [CrossRef]
- Kulkova, I.; Wróbel, B.; Dobrzyński, J. Serratia Spp. as Plant Growth-Promoting Bacteria Alleviating Salinity, Drought, and Nutrient Imbalance Stresses. Front. Microbiol. 2024, 15, 1342331. [Google Scholar] [CrossRef]
- Desoky, E.-S.M.; Saad, A.M.; El-Saadony, M.T.; Merwad, A.-R.M.; Rady, M.M. Plant Growth-Promoting Rhizobacteria: Potential Improvement in Antioxidant Defense System and Suppression of Oxidative Stress for Alleviating Salinity Stress in Triticum aestivum (L.) Plants. Biocatal. Agric. Biotechnol. 2020, 30, 101878. [Google Scholar] [CrossRef]
- Takarada, H.; Sekine, M.; Kosugi, H.; Matsuo, Y.; Fujisawa, T.; Omata, S.; Kishi, E.; Shimizu, A.; Tsukatani, N.; Tanikawa, S.; et al. Complete Genome Sequence of the Soil Actinomycete Kocuria rhizophila. J. Bacteriol. 2008, 190, 4139–4146. [Google Scholar] [CrossRef]
- Ningsih, F.; Sari, D.C.a.F.; Yabe, S.; Yokota, A.; Oetari, A.; Sjamsuridzal, W. Antibacterial Activity of Microbispora rosea Subsp. Rosea SL3-2-R-1 Grown on Different Media and Solidifying Agents. J. Phys. Conf. Ser. 2021, 1918, 052011. [Google Scholar] [CrossRef]
- Al-Karablieh, N.; Mutlak, I.; Al-Dokh, A. Isolation and Identification of Pseudomonas viridiflava, the Causal Agent of Fruit Rotting of Cucumis sativus. Jordan J. Agric. Sci. 2017, 13, 79–91. [Google Scholar] [CrossRef]
- Lipps, S.M.; Samac, D.A. Pseudomonas viridiflava: An Internal Outsider of the Pseudomonas syringae Species Complex. Mol. Plant Pathol. 2021, 23, 3–15. [Google Scholar] [CrossRef]
- Sarris, P.F.; Trantas, E.A.; Mpalantinaki, E.; Ververidis, F.; Goumas, D.E. Pseudomonas viridiflava, a Multi Host Plant Pathogen with Significant Genetic Variation at the Molecular Level. PLoS ONE 2012, 7, e36090. [Google Scholar] [CrossRef]
- Alimi, M.; Rahimian, H.; Hassanzadeh, N.; Darzi, M.T.; Ahmadikhah, A.; Heydari, A.; Balestra, G.M. First Detection of Pseudomonas viridiflava, the Causal Agent of Blossom Blight in Apple by Using Specific Designed Primers. Afr. J. Microbiol. Res. 2011, 5, 4708–4713. [Google Scholar] [CrossRef]
- Hasan, M.F.; Islam, M.A.; Sikdar, B. First Report of Serratia Marcescens Associated with Black Rot of Citrus Sinensis Fruit, and Evaluation of Its Biological Control Measures in Bangladesh. F1000Research 2022, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Jha, P.N. The Multifarious PGPR Serratia Marcescens CDP-13 Augments Induced Systemic Resistance and Enhanced Salinity Tolerance of Wheat (Triticum aestivum L.). PLoS ONE 2016, 11, e0155026. [Google Scholar] [CrossRef]
- Hamada, M.A.; Soliman, E.R.S. Characterization and Genomics Identification of Key Genes Involved in Denitrification-DNRA-Nitrification Pathway of Plant Growth-Promoting Rhizobacteria (Serratia Marcescens OK482790). BMC Microbiol. 2023, 23, 210. [Google Scholar] [CrossRef]
- Dogra, N.; Yadav, R.; Kaur, M.; Adhikary, A.; Kumar, S.; Ramakrishna, W. Nutrient Enhancement of Chickpea Grown with Plant Growth Promoting Bacteria in Local Soil of Bathinda, Northwestern India. Physiol. Mol. Biol. Plants 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Gyaneshwar, P.; James, E.K.; Mathan, N.; Reddy, P.M.; Reinhold-Hurek, B.; Ladha, J.K. Endophytic Colonization of Rice by a Diazotrophic Strain of Serratia marcescens. J. Bacteriol. 2001, 183, 2634–2645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Z.; Zhang, M.; Li, X.; Wang, M.; Li, L.; Li, X.; Ding, Z.; Tian, H. Serratia marcescens PLR Enhances Lateral Root Formation Through Supplying PLR-Derived Auxin and Enhancing Auxin Biosynthesis in Arabidopsis. J. Exp. Bot. 2022, 73, 3711–3725. [Google Scholar] [CrossRef]
- Ramos, P.L.; Trappen, S.V.; Thompson, F.L.; Rocha, R.C.S.; Barbosa, H.R.; Vos, P.D.; Moreira-Filho, C.A. Screening for Endophytic Nitrogen-Fixing Bacteria in Brazilian Sugar Cane Varieties Used in Organic Farming and Description of Stenotrophomonas pavanii Sp. Nov. Int. J. Syst. Evol. Microbiol. 2011, 61, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wu, X.; Li, C.; Zhang, X.; Wang, P.; Tan, X.; Liu, Y.; Zhang, D.; Chen, Y. Analysis of the Antagonistic Effect of Stenotrophomonas geniculata WXY53 on Magnaporthe oryzae Through Bioassays and Whole-Genome Sequencing. Biol. Control 2024, 196, 105587. [Google Scholar] [CrossRef]
- CABI Head Office. Burkholderia andropogonis (Bacterial Leaf Stripe of Sorghum and Corn). PlantwisePlus Knowledge Bank 2022, Species Compendium, doi:10.1079/pwkb.species.44927. Available online: https://plantwiseplusknowledgebank.org/doi/full/10.1079/pwkb.species.44927 (accessed on 10 March 2025).
- Cother, E.J.; Noble, D.; Peters, B.J.; Albiston, A.; Ash, G.J. A New Bacterial Disease of Jojoba (Simmondsia chinensis) Caused by Burkholderia Andropogonis. Plant Pathol. 2004, 53, 129–135. [Google Scholar] [CrossRef]
- Chen, J.; Lin, J.; Gan, Y.; Tong, Z.; Han, X.; Duan, X.; Gan, B.; Yan, J. First Report of Sphingobacterium multivorum Causing Bacterial Rot of Cauliflower Mushroom (Sparassis latifolia) in China. Plant Dis. 2024, 108, 1881. [Google Scholar] [CrossRef]
- Wang, Y.; Brons, J.K.; van Elsas, J.D. Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum. Microorganisms 2021, 9, 2057. [Google Scholar] [CrossRef]
- Padder, S.A.; Mansoor, S.; Bhat, S.A.; Baba, T.R.; Rather, R.A.; Wani, S.M.; Popescu, S.M.; Sofi, S.; Aziz, M.A.; Hefft, D.I.; et al. Bacterial Endophyte Community Dynamics in Apple (Malus domestica Borkh.) Germplasm and Their Evaluation for Scab Management Strategies. Journal of Fungi. 2021, 7, 923. [Google Scholar] [CrossRef]
- Lim, H.S.; Kim, Y.S.; Kim, S.D. Pseudomonas stutzeri YPL-1 Genetic Transformation and Antifungal Mechanism Against Fusarium solani, an Agent of Plant Root Rot. Appl. Environ. Microbiol. 1991, 57, 510–516. [Google Scholar] [CrossRef]
- Zhao, Y.; Ding, W.-J.; Xu, L.; Sun, J.Q. A comprehensive comparative genomic analysis revealed that plant growth promoting traits are ubiquitous in strains of Stenotrophomonas. Front. Microbiol. 2024, 15, 1395477. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.S.; Moffat, C.S.; Lopez-Ruiz, F.J.; Gibberd, M.R.; Hamblin, J.; Zerihun, A. Host–multi-pathogen warfare: Pathogen interactions in co-infected plants. Front. Plant Sci. 2017, 8, 1806. [Google Scholar] [CrossRef] [PubMed]
- Binyamin, R.; Nadeem, S.M.; Akhtar, S.; Khan, M.Y.; Anjum, R. Beneficial and pathogenic plant-microbe interactions: A review. Soil Environ. 2019, 38, 127–150. [Google Scholar] [CrossRef]
- Haney, C.H.; Samuel, B.S.; Bush, J.; Ausubel, F.M. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat. Plants 2015, 1, 15051. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Kazhydromet Climatic Data for Kazakhstan, 2020–2024. Available online: http://ecodata.kz:3838/dm_climat_ru/ (accessed on 12 May 2025).
- Deakin, G.; Tilston, E.L.; Bennett, J.; Passey, T.; Harrison, N.; Fernández-Fernández, F.; Xu, X. Spatial structuring of soil microbial communities in commercial apple orchards. Appl. Soil Ecol. 2018, 130, 1–12. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Site | Coordinates | Altitude (m.a.s.l.) | Number of Samples |
|---|---|---|---|---|
| Zhongar Alatau | Phenological site | N 45.51963° E 80.73385° | 1400 | 57 |
| Genetic reserve of Siever’s wild apple trees (Zhng) | N 45.51746° E 80.72224° | 1255 | 52 | |
| Ile Alatau | Tau-Turgen | N 43.364244° E 77.680405° | 1300 | 55 |
| Genetic reserve of Siever’s wild apple trees (Ile) | N 43.36681° E 77.67301° | 1585 | 70 | |
| Ketmen | Sumbe | N 43.293056° E 79.484167° | 1734 | 50 |
| Ketpentau | N 43.305278° E 79.751389° | 1845 | 50 |
| Mountain Ranges | Number of Processed Reads | Percentage of Classified Reads | Mean Read Length (bp) | Mean Read Quality (PHRED) | N50 | Total Bases (Megabases) |
|---|---|---|---|---|---|---|
| Zhongar Alatau | 18,549 | 100% | 1426.8 | 12.2 | 1461.0 | 26.5 Mb |
| Ile Alatau | 71,859 | 99.94% | 1327.4 | 12.2 | 1441.0 | 95 Mb |
| Ketmen | 58,325 | 99.99% | 1341.9 | 12.2 | 1449.0 | 78 Mb |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendybayeva, A.; Makhambetov, A.; Yanin, K.; Taskuzhina, A.; Khusnitdinova, M.; Gritsenko, D. Metagenomic Analysis of Wild Apple (Malus sieversii) Trees from Natural Habitats of Kazakhstan. Plants 2025, 14, 1511. https://doi.org/10.3390/plants14101511
Mendybayeva A, Makhambetov A, Yanin K, Taskuzhina A, Khusnitdinova M, Gritsenko D. Metagenomic Analysis of Wild Apple (Malus sieversii) Trees from Natural Habitats of Kazakhstan. Plants. 2025; 14(10):1511. https://doi.org/10.3390/plants14101511
Chicago/Turabian StyleMendybayeva, Aruzhan, Alibek Makhambetov, Kirill Yanin, Aisha Taskuzhina, Marina Khusnitdinova, and Dilyara Gritsenko. 2025. "Metagenomic Analysis of Wild Apple (Malus sieversii) Trees from Natural Habitats of Kazakhstan" Plants 14, no. 10: 1511. https://doi.org/10.3390/plants14101511
APA StyleMendybayeva, A., Makhambetov, A., Yanin, K., Taskuzhina, A., Khusnitdinova, M., & Gritsenko, D. (2025). Metagenomic Analysis of Wild Apple (Malus sieversii) Trees from Natural Habitats of Kazakhstan. Plants, 14(10), 1511. https://doi.org/10.3390/plants14101511