
Integrative Analysis of Transcriptomic and Metabolomic Profiles Uncovers the Mechanism of Color Variation in the Tea Plant Callus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
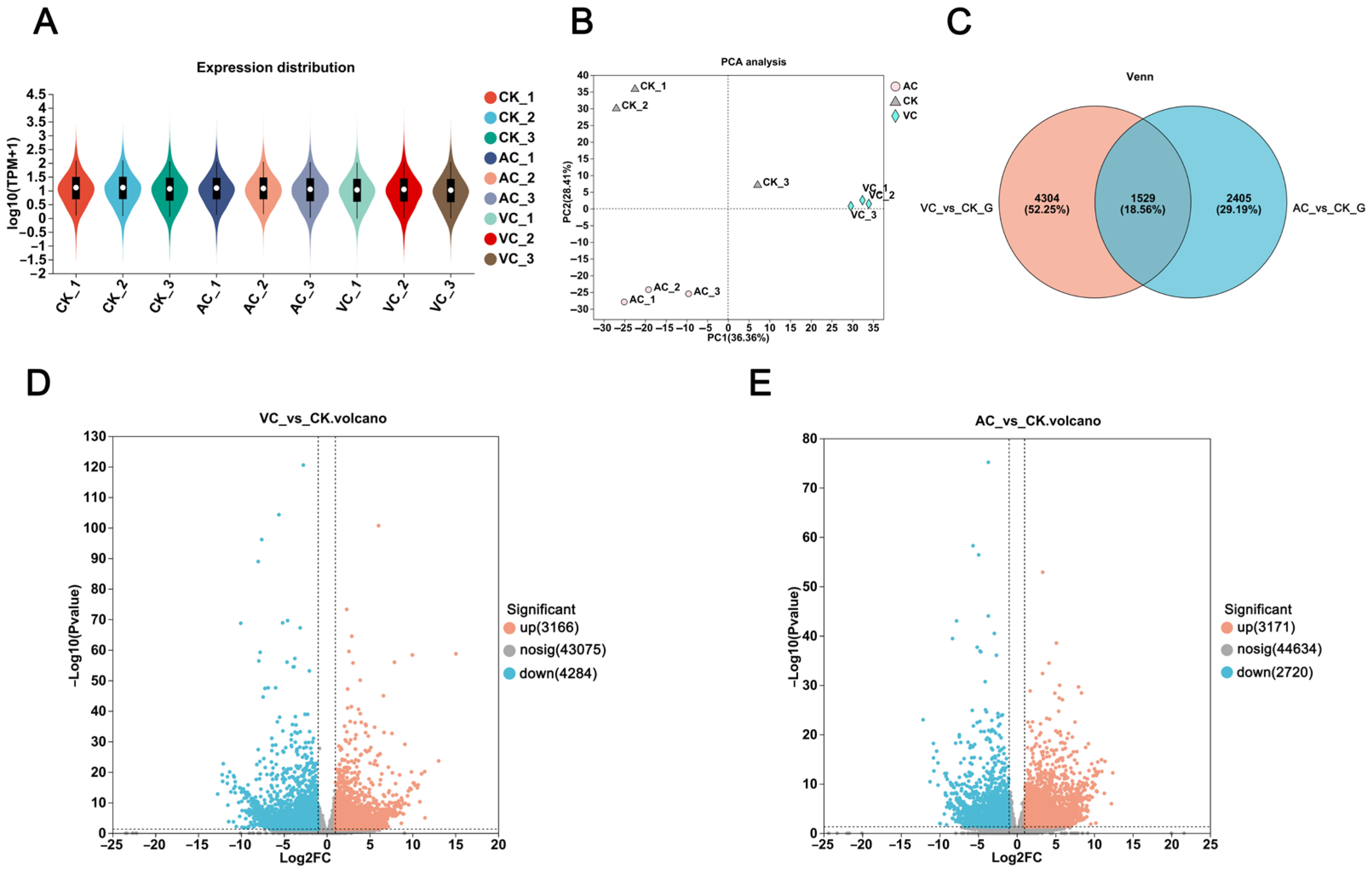
2.1. Analysis of Differentially Expressed Genes (DEGs)
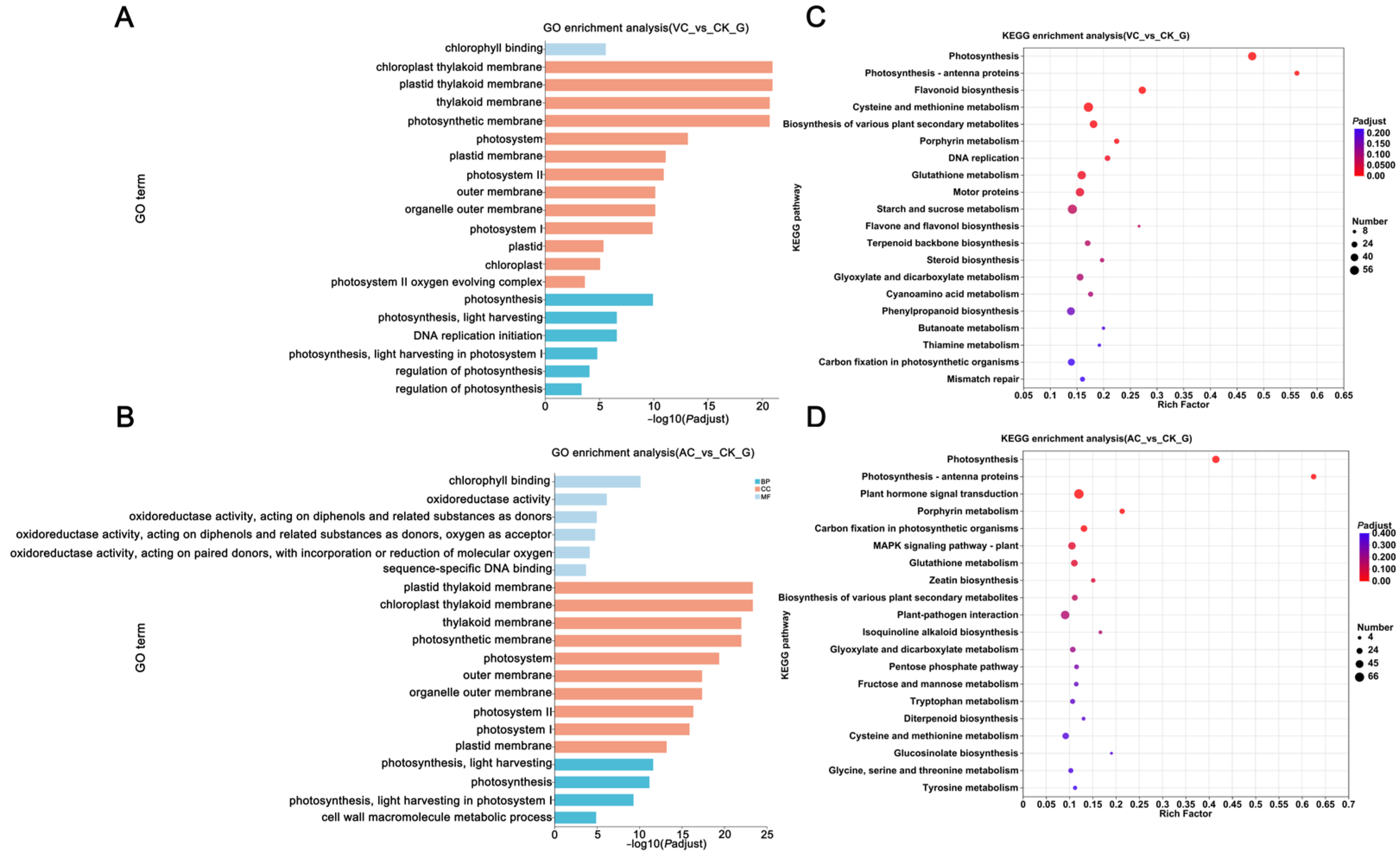
2.2. Enrichment Analysis
2.3. Analysis of DEGs Related to Photosynthesis-Related Synthetic Pathways
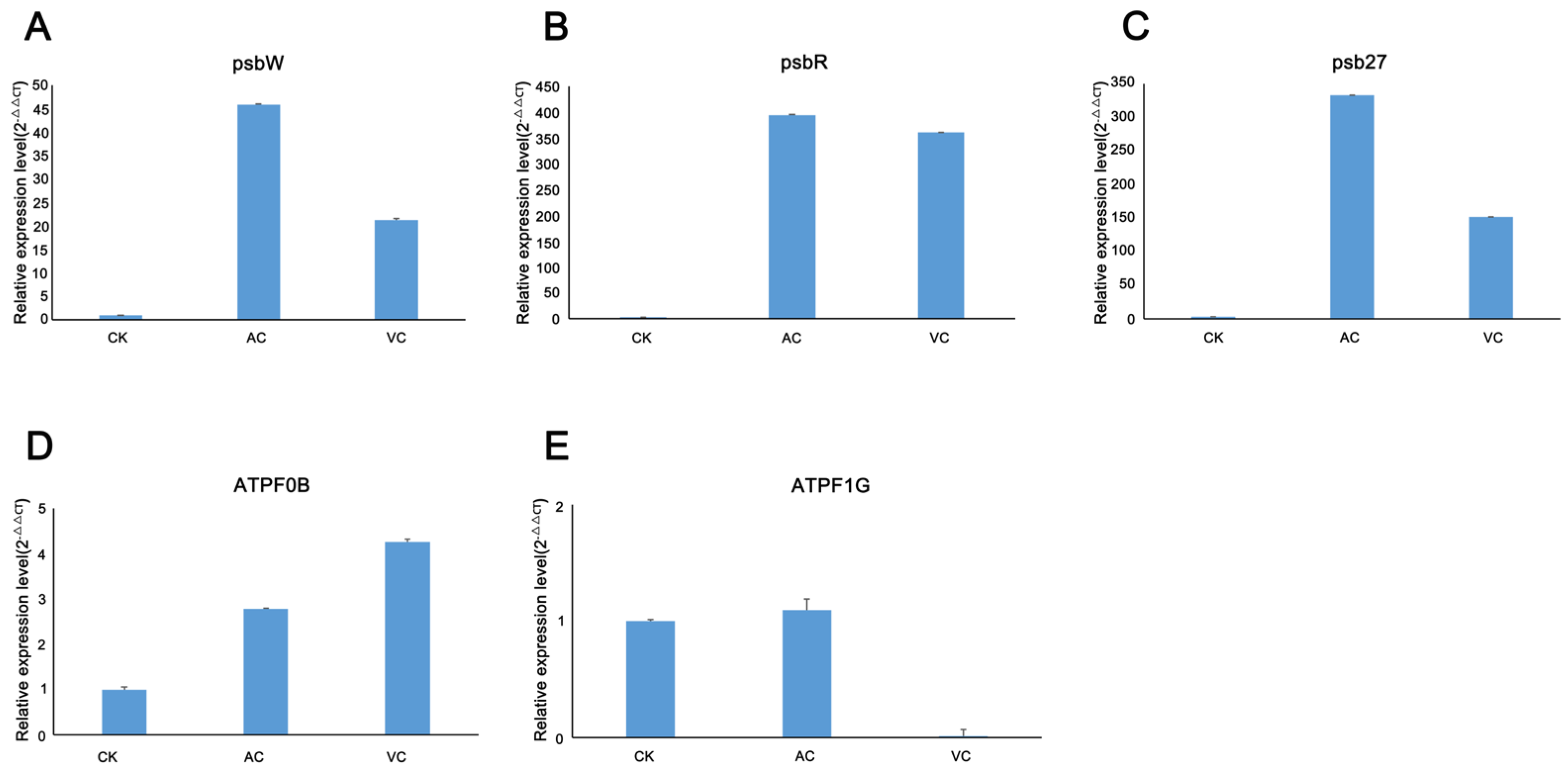
2.4. Relative Expression Analysis of Selected Genes in Photosynthesis Process
2.5. Metabolite Profiling of Callus Tissues with Different Colors
2.6. Enrichment Analysis of Differential Metabolites (DEMs)
2.7. Integrated Analysis of Transcriptome and Metabolome
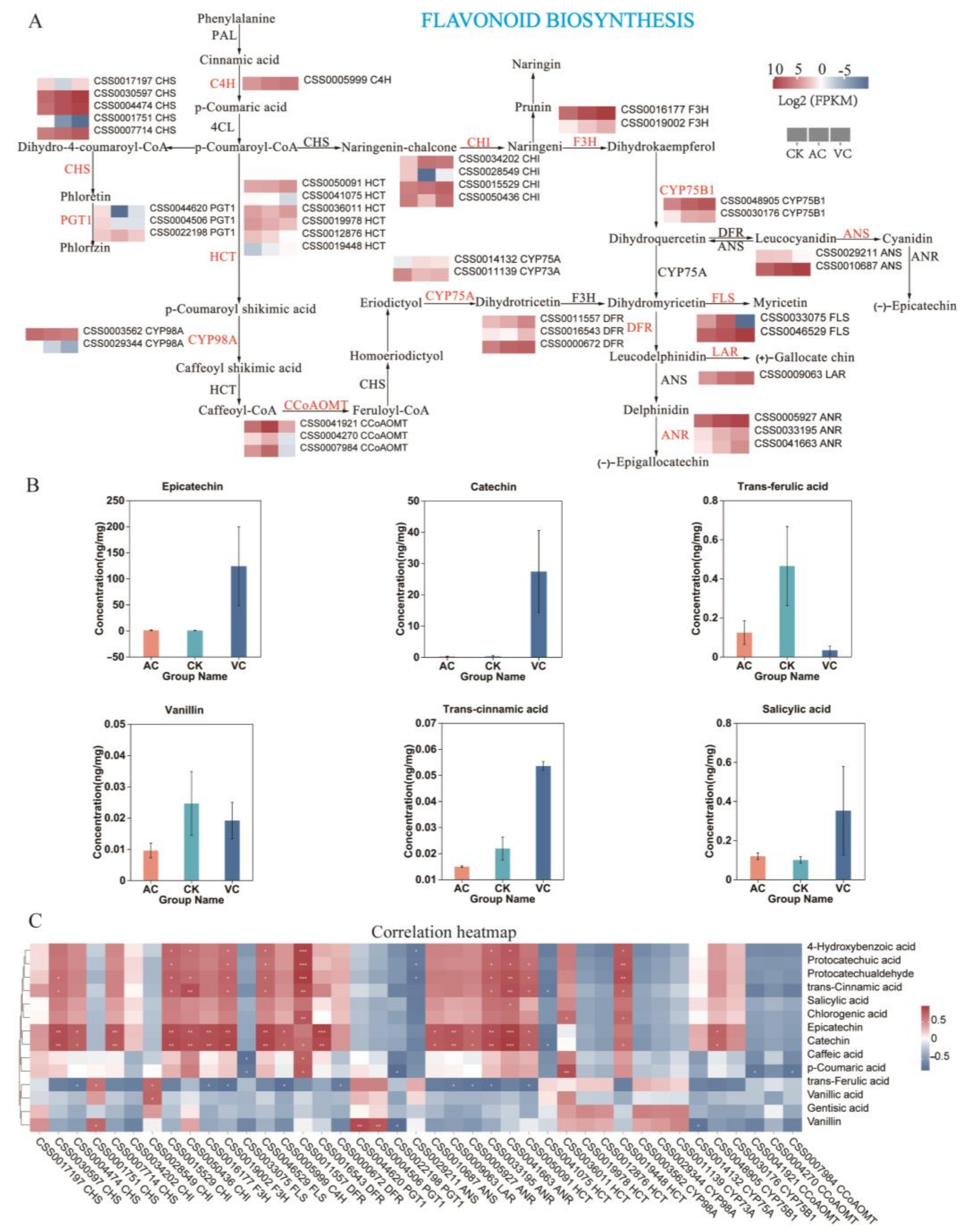
2.8. Flavonoid Biosynthesis Pathway
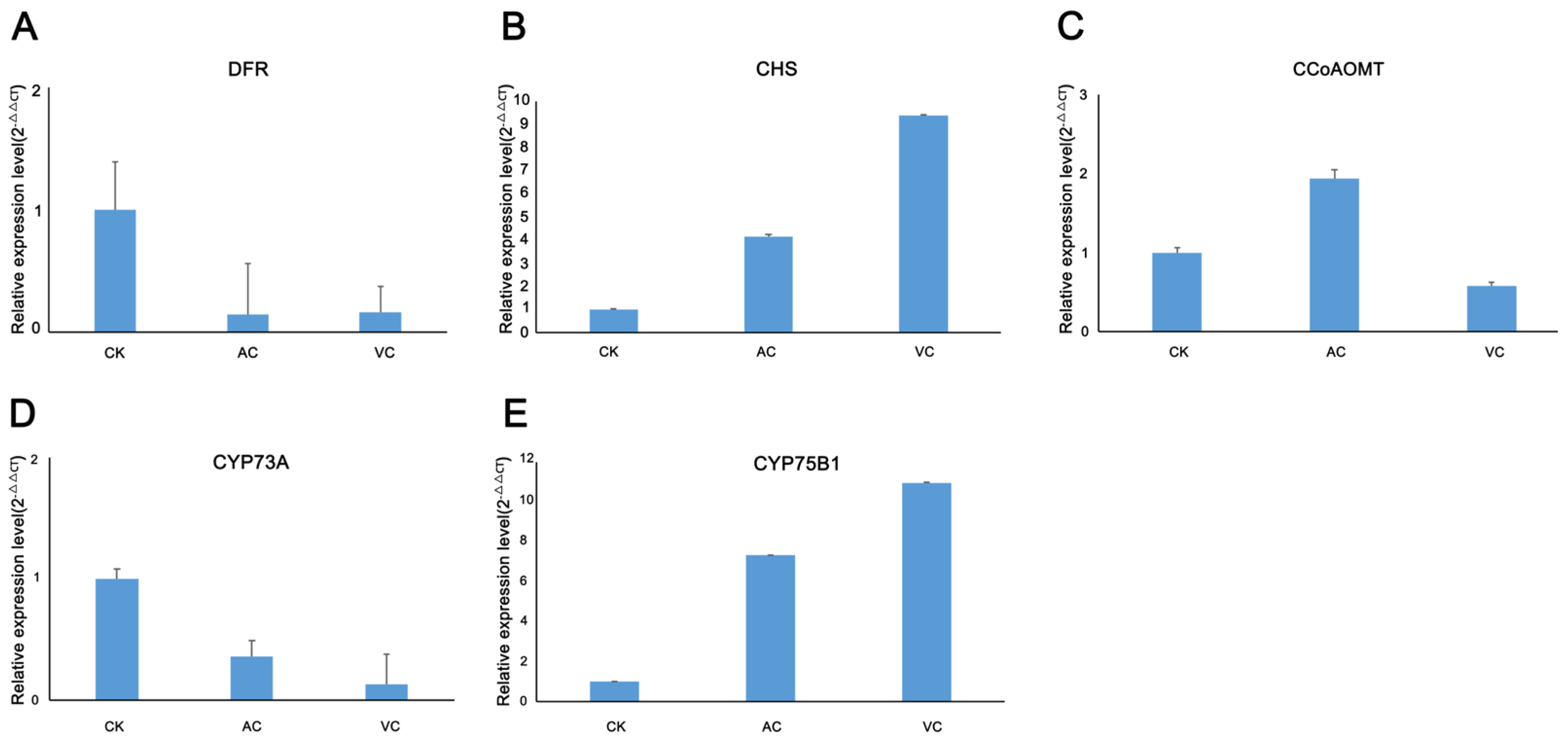
2.9. Validation of Relative Expression of Selected Genes in the Flavonoid Biosynthesis Pathway via qRT-PCR
3. Discussion
3.1. Differences in Metabolite Accumulation Affecting Callus Color Change
3.2. Anti-Browning Agent-Activated Carbon Maintains Green Phenotypes via Enhanced Photosynthesis
3.3. Ascorbic Acid Modulates Pigment Formation in Tea Callus by Regulating Key Gene Expression
4. Materials and Methods
4.1. Experimental Materials
4.2. Library Construction and Transcriptome Sequencing
4.3. Metabolome Detection
4.4. Validation via qRT-PCR
4.5. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lan, Z.; Shen, X. Lancang River: The Cradle of Tea Civilization; China Forestry Publishing House: Beijing, China, 2021. [Google Scholar]
- Wang, F.; Feng, H.; Zheng, Y.; Liu, R.; Dong, J.; Wu, Y.; Chen, S.; Zhang, B.; Wang, P.; Yan, J. Aroma analysis and biomarker screening of 27 tea cultivars based on four leaf color types. Food Res. Int. 2025, 201, 115681. [Google Scholar] [CrossRef]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial effects of green tea: A literature review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fu, J.; Xu, Y.; Shen, Y.; Zhang, Y.; Ye, Z.; Tong, W.; Zeng, X.; Yang, J.; Tang, D.; et al. CsMYB1 integrates the regulation of trichome development and catechins biosynthesis in Camellia sinensis domestication. New Phytol. 2022, 234, 902–917. [Google Scholar] [CrossRef]
- Qiu, H.; Zhang, X.; Zhang, Y.; Jiang, X.; Ren, Y.; Gao, D.; Zhu, X.; Usadel, B.; Fernie, A.R.; Wen, W. Depicting the genetic and metabolic panorama of chemical diversity in the Camellia sinensis. Plant Biotechnol. J. 2024, 22, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Verma, T.; Sinha, M.; Bansal, N.; Yadav, S.R.; Shah, K.; Chauhan, N.S. Plants Used as Antihypertensive. Nat. Prod Bioprospect. 2021, 11, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Qi, B.; Mou, C.; Wang, L.; Jiao, Y.; Dou, Y.; Zheng, H. BREVIPEDICELLUS and ERECTA control the expression of AtPRX17 to prevent Arabidopsis callus browning. J. Exp. Bot. 2022, 73, 1516–1532. [Google Scholar] [CrossRef]
- Wu, K.; Liu, Y.; Xu, Y.; Yu, Z.; Cao, Q.; Gong, H.; Yang, Y.; Ye, J.; Jia, X. Unveiling the Molecular Mechanisms of Browning in Camellia hainanica Callus through Transcriptomic and Metabolomic Analysis. Int. J. Mol. Sci. 2024, 25, 11021. [Google Scholar] [CrossRef]
- Ciou, J.Y.; Lin, H.H.; Chiang, P.Y.; Wang, C.C.; Charles, A.L. The role of polyphenol oxidase and peroxidase in the browning of water caltrop pericarp during heat treatment. Food Chem. 2011, 127, 523–527. [Google Scholar] [CrossRef]
- He, Y.; Guo, X.; Lu, R.; Niu, B.; Pasapula, V.; Hou, P.; Cai, F.; Xu, Y.; Chen, F. Changes in morphology and biochemical indices in browning callus derived from Jatropha curcas hypocotyls. Plant Cell Tissue Organ Cult. 2009, 98, 11–17. [Google Scholar] [CrossRef]
- Rana, M.M.; Han, Z.X.; Song, D.P.; Liu, G.F.; Li, D.X.; Wan, X.C.; Karthikeyan, A.; Wei, S. Effect of Medium Supplements on Agrobacterium rhizogenes Mediated Hairy Root Induction from the Callus Tissues of Camellia sinensis var. sinensis. Int. J. Mol. Sci. 2016, 17, 1132. [Google Scholar] [CrossRef]
- Aksenova, M.A.; Nechaeva, T.L.; Goncharuk, E.A.; Zubova, M.Y.; Kazantseva, V.V.; Lapshin, P.V.; Frolov, A.; Zagoskina, N.V. Changes in the Antioxidant Potential of Camellia sinensis Cultures under the Influence of Phenolic Precursors. Molecules 2024, 29, 474. [Google Scholar] [CrossRef]
- Alagarsamy, K.; Shamala, L.F.; Wei, S. Influence of media supplements on inhibition of oxidative browning and bacterial endophytes of Camellia sinensis var. sinensis. 3 Biotech 2018, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.D. The role of activated charcoal in plant tissue culture. Biotechnol. Adv. 2008, 26, 618–631. [Google Scholar]
- Wen, Y.T.; Liang, Y.Q.; Chai, W.M.; Wei, Q.M.; Yu, Z.Y.; Wang, L.J. Effect of ascorbic acid on tyrosinase and its anti-browning activity in fresh-cut Fuji apple. J. Food Biochem. 2021, 45, e13995. [Google Scholar] [CrossRef]
- Guan, Y.; Lu, S.; Sun, Y.; Zheng, X.; Wang, R.; Lu, X.; Pang, L.; Cheng, J.; Wang, L. Tea Polyphenols Inhibit the Occurrence of Enzymatic Browning in Fresh-Cut Potatoes by Regulating Phenylpropanoid and ROS Metabolism. Plants 2024, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Xu, W.; Zhang, L.; Shi, X.; Zhou, J.; Liu, H.; Zhu, K. Integrated Metabolomic and Transcriptomic Analyses Reveal the Potential Molecular Mechanism Underlying Callus Browning in Paeonia ostii. Plants 2025, 14, 560. [Google Scholar] [CrossRef]
- Ma, K.F.; Zhang, Q.X.; Cheng, T.R.; Yan, X.L.; Pan, H.T.; Wang, J. Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 2315. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Wang, Z.; Yang, P. Review: The effect of light on the key pigment compounds of photosensitive etiolated Camellia sinensis. Bot. Stud. 2021, 62, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, C.F.; Ma, J.Q.; Huang, D.J.; Ma, C.L.; Jin, J.Q.; Yao, M.Z.; Chen, L. Comprehensive Dissection of Metabolic Changes in Albino and Green Tea Cultivars. J. Agric Food Chem. 2018, 66, 2040–2048. [Google Scholar] [CrossRef]
- Huang, Q.; Jia, H.; Wang, H. Clones of Portulaca oleracea and their plant regeneration system. North. Hortic. 2007, 2, 142–145. [Google Scholar]
- Li, N.N.; Lu, J.L.; Li, Q.S.; Zheng, X.Q.; Wang, X.C.; Wang, L.; Wang, Y.C.; Ding, C.Q.; Liang, Y.R.; Yang, Y.J. Dissection of Chemical Composition and Associated Gene Expression in the Pigment-Deficient Tea Cultivar ‘Xiaoxueya’ Reveals an Albino Phenotype and Metabolite Formation. Front Plant Sci. 2019, 27, 10. [Google Scholar] [CrossRef]
- Kallscheuer, N.; Vogt, M.; Marienhagen, J. A Novel Synthetic Pathway Enables Microbial Production of Polyphenols Independent from the Endogenous Aromatic Amino Acid Metabolism. ACS Synth. Biol. 2017, 6, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. J. 2001, 126, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zhang, W.; Liu, Y.; Zhang, H.; Ren, H.; Chen, Y.; Wang, L.; Zeng, J.; Yang, Y.; Wang, X. Pale green mutant analyses reveal the importance of CsGLKs in chloroplast developmental regulation and their effects on flavonoid biosynthesis in Camellia sinensis. Plant Physiol. Biochem. 2020, 146, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ming, H.; Xi, Z.; Fang, Z.; Cheng, W.; Zhong, H. Multi-omics research in albino Camellia sinensis: Past, present, and future. Sci. Hortic. 2020, 261, 108943. [Google Scholar]
- Zhu, M.Z.; Zhou, F.; Ran, L.S.; Li, Y.L.; Tan, B.; Wang, K.B.; Huang, J.A.; Liu, Z.H. Metabolic Profiling and Gene Expression Analyses of Purple-Leaf Formation in Tea Cultivars (Camellia sinensis var. Sinensis and var. assamica). Front. Plant Sci. 2021, 12, 606962. [Google Scholar] [CrossRef]
- Chen, X.; Wang, P.; Zheng, Y.; Gu, M.; Lin, X.; Wang, S.; Jin, S.; Ye, N. Comparison of Metabolome and Transcriptome of Flavonoid Biosynthesis Pathway in a Purple-Leaf Tea Germplasm Jinmingzao and a Green-Leaf Tea Germplasm Huangdan reveals Their Relationship with Genetic Mechanisms of Color Formation. Int. J. Mol. Sci. 2020, 21, 4167. [Google Scholar] [CrossRef]
- Jarvis, P.; López-Juez, E. Biogenesis and homeostasis of chloroplasts and other plastids. Nat. Rev. Mol. Cell Biol. 2013, 14, 787–802. [Google Scholar]
- Jin, J.; Chen, Y.; Cai, J.; Lv, L.; Zeng, X.; Li, J.; Asghar, S.; Li, Y. Establishment of an efficient regeneration system of ‘ZiKui’ tea with hypocotyl as explants. Sci Rep. 2024, 21, 14. [Google Scholar] [CrossRef]
- Pace, B.; Capotorto, I.; Palumbo, M.; Pelosi, S.; Cefola, M. Combined Effect of Dipping in Oxalic or in Citric Acid and Low O Modified Atmosphere, to Preserve the Quality of Fresh-Cut Lettuce during Storage. Foods 2020, 9, 988. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, M.; Liu, J.; Cai, J. Metabolomic and Transcriptomic Analyses Reveal the Characteristics of Tea Flavonoids and Caffeine Accumulation and Regulation between Chinese Varieties (Camellia sinensis var. sinensis) and Assam Varieties (C. sinensis var. assamica). Genes 2022, 13, 1994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, Z.; Wang, Y.; Zhang, X.; Kong, W. Haplotype-Resolution Transcriptome Analysis Reveals Important Responsive Gene Modules and Allele-Specific Expression Contributions under Continuous Salt and Drought in Camellia sinensis. Genes 2023, 14, 1417. [Google Scholar] [CrossRef] [PubMed]
- Ntezimana, B.; Xu, W.; Li, Y.; Zhou, J.; Pathak, S.; Chen, Y.; Yu, Z.; Zhang, D.; Ni, D. Integrated Transcriptomic and Metabolomic Analyses Reveal Changes in Aroma- and Taste-Related Substances During the Withering Process of Black Tea. Foods. 2024, 13, 3977. [Google Scholar] [PubMed]
- Zubova, M.Y.; Goncharuk, E.A.; Nechaeva, T.L.; Aksenova, M.A.; Zaitsev, G.P.; Katanskaya, V.M.; Kazantseva, V.V.; Zagoskina, N.V. Influence of Primary Light Exposure on the Morphophysiological Characteristics and Phenolic Compounds Accumulation of a Tea Callus Culture (Camellia sinensis L.). Int. J. Mol. Sci. 2024, 25, 10420. [Google Scholar] [CrossRef]
- Wan, X.; Wu, J.; Wang, X.; Cui, L.; Xiao, Q. Accumulation patterns of flavonoids and phenolic acids in different colored sweet potato flesh revealed based on untargeted metabolomics. Food Chem. X 2024, 23, 101551. [Google Scholar] [CrossRef]
- Aksenova, M.A.; Nechaeva, T.L.; Zubova, M.Y.; Goncharuk, E.A.; Kazantseva, V.V.; Katanskaya, V.M.; Lapshin, P.V.; Zagoskina, N.V. Influence of Different Precursors on Content of Polyphenols in Camellia sinensis Vitro Callus Culture. Plants 2023, 12, 796. [Google Scholar] [CrossRef]
- Wu, H.; Haig, T.; Pratley, J.; Lemerle, D.; An, M. Allelochemicals in wheat (Triticum aestivum L.): Variation of phenolic acids in shoot tissues. J. Chem. Ecol. 2001, 27, 125–135. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Wu, J.; Lv, S.; Zhao, L.; Gao, T.; Yu, C.; Hu, J.; Ma, F. Advances in the study of the function and mechanism of the action of flavonoids in plants under environmental stresses. Planta 2023, 257, 108. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Mazaheri-Tirani, M.; Parsa Motlagh, B.; Ahmadzadeh, M.; Seyedi, A. Mitigating pb toxicity in Sesbania sesban L. through activated charcoal supplementation: A hydroponic study on enhanced phytoremediation. BMC. Plant Biol. 2024, 24, 744. [Google Scholar] [CrossRef] [PubMed]
- Fristedt, R.; Williams-Carrier, R.; Merchant, S.S.; Barkan, A. A thylakoid membrane protein harboring a DnaJ-type zinc finger domain is required for photosystem I accumulation in plants. J. Biol. Chem. 2014, 289, 30657–30667. [Google Scholar] [CrossRef] [PubMed]
- Rühle, T.; Leister, D.; Pasch, V. Chloroplast ATP synthase: From structure to engineering. Plant Cell 2024, 36, 3974–3996. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, A.N. pH-dependent regulation of electron transport and ATP synthesis in chloroplasts. Photosynth. Res. 2013, 116, 511–534. [Google Scholar] [CrossRef]
- Nam, H.I.; Shahzad, Z.; Dorone, Y.; Clowez, S.; Zhao, K.; Bouain, N.; Lay-Pruitt, K.S.; Cho, H.; Rhee, S.Y.; Rouached, H. Interdependent iron and phosphorus availability controls photosynthesis through retrograde signaling. Nat. Commun. 2021, 12, 7211. [Google Scholar] [CrossRef]
- Wang, Y.W.; Chen, S.M.; Wang, W.J.; Huang, X.Q.; Zhou, C.F.; Zhuang, Z.; Lu, S. The DnaJ-Like Zinc Finger Domain Protein PSA2 Affects Light Acclimation and Chloroplast Development in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 360. [Google Scholar] [CrossRef]
- Sun, X.; He, L.; Guo, Z.; Xiao, Z.; Su, J.; Liu, X.; Zhou, H.; Li, C.; Gao, H. Comparative transcriptome analyses reveal genes related to pigmentation in the petals of a flower color variation cultivar of Rhododendron obtusum. Mol. Biol. Rep. 2022, 49, 2641–2653. [Google Scholar] [CrossRef]
- Zou, Q.; Bao, T.; Yu, L.; Xu, H.; Liu, W.; Li, Z.; Zhu, Y.; Chen, R.; Hou, X.; Zhang, Z.; et al. The regulatory module MdCPCL-MdILR3L mediates the synthesis of ascorbic acid and anthocyanin in apple. Plant Biotechnol. J. 2025, 23, 1101–1117. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar]
- Si, C.; Dong, W.; Teixeira, D.; Jaime, A.; He, C.; Yu, Z.; Zhang, M.; Huang, L.; Zhao, C.; Zeng, D.; et al. Functional analysis of flavanone 3-hydroxylase (F3H) from Dendrobium officinale, which confers abiotic stress tolerance. Hortic. Plant J. 2023, 9, 356–364. [Google Scholar] [CrossRef]
- Zhao, T.; Wei, Z.; Liu, H.; Dong, L. Integrated Metabolome and Transcriptome Analyses Reveal That the Flavonoid Metabolic Pathway Is Associated with Pigment Differential Accumulation in Two Colors of Petaloid Staminodes in Canna glauca. Horticulturae 2024, 10, 372. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Li, X.; Liu, Z.; Li, F.; Huang, H.; Huang, M. Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa. Horticulturae 2024, 10, 415. [Google Scholar] [CrossRef]
- Bassolino, L.; Petroni, K.; Polito, A.; Marinelli, A.; Azzini, E.; Ferrari, M.; Ficco, D.B.M.; Mazzucotelli, E.; Tondelli, A.; Fricano, A.; et al. Does Plant Breeding for Antioxidant-Rich Foods Have an Impact on Human Health? Antioxidants 2022, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fan, Z.; Tong, R.; Zhou, X.; Li, J.; Yin, H. Functional Diversification of the Dihydroflavonol 4-Reductase from Camellia nitidissima Chi. in the Control of Polyphenol Biosynthesis. Genes 2020, 11, 1341. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, N.; Lee, C.H.; Wong, S.L.; Fauzi, C.E.N.C.A.; Zamri, N.M.A.; Lee, T.H. Prevention of Enzymatic Browning by Natural Extracts and Genome-Editing: A Review on Recent Progress. Molecules 2022, 27, 1101. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Min, D.; Yi, L.; Chun, W.; Xiao, Y.; Huang Ren, H.; Huang Qi, H.; Shi, Z. Transcriptome and targeted metabolome analysis of regulatory factors of carotenoids in pepper fruits. Acta Hortic. Sin. 2024, 51, 1773–1791. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Liangsheng, Z.; Pedersen, B.S.; Fidel, R.; Alex, W.V.; Aurélien, N.; Mungall, C.J.; Yunes, J.M.; Olga, B.; Mark, W. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Tian, Y.; Wang, Y.; Guan, Y.; Zhang, Y.; Zhang, Y.; Tao, Y.; Lan, Z.; Wang, D. Integrative Analysis of Transcriptomic and Metabolomic Profiles Uncovers the Mechanism of Color Variation in the Tea Plant Callus. Plants 2025, 14, 1454. https://doi.org/10.3390/plants14101454
Xiao M, Tian Y, Wang Y, Guan Y, Zhang Y, Zhang Y, Tao Y, Lan Z, Wang D. Integrative Analysis of Transcriptomic and Metabolomic Profiles Uncovers the Mechanism of Color Variation in the Tea Plant Callus. Plants. 2025; 14(10):1454. https://doi.org/10.3390/plants14101454
Chicago/Turabian StyleXiao, Mengna, Yingju Tian, Ya Wang, Yunfang Guan, Ying Zhang, Yuan Zhang, Yanlan Tao, Zengquan Lan, and Dexin Wang. 2025. "Integrative Analysis of Transcriptomic and Metabolomic Profiles Uncovers the Mechanism of Color Variation in the Tea Plant Callus" Plants 14, no. 10: 1454. https://doi.org/10.3390/plants14101454
APA StyleXiao, M., Tian, Y., Wang, Y., Guan, Y., Zhang, Y., Zhang, Y., Tao, Y., Lan, Z., & Wang, D. (2025). Integrative Analysis of Transcriptomic and Metabolomic Profiles Uncovers the Mechanism of Color Variation in the Tea Plant Callus. Plants, 14(10), 1454. https://doi.org/10.3390/plants14101454