
Transcriptome-Wide N6-Methyladenosine (m6A) Methylation Analyses in a Compatible Wheat–Puccinia striiformis f. sp. tritici Interaction



Abstract
1. Introduction
2. Results
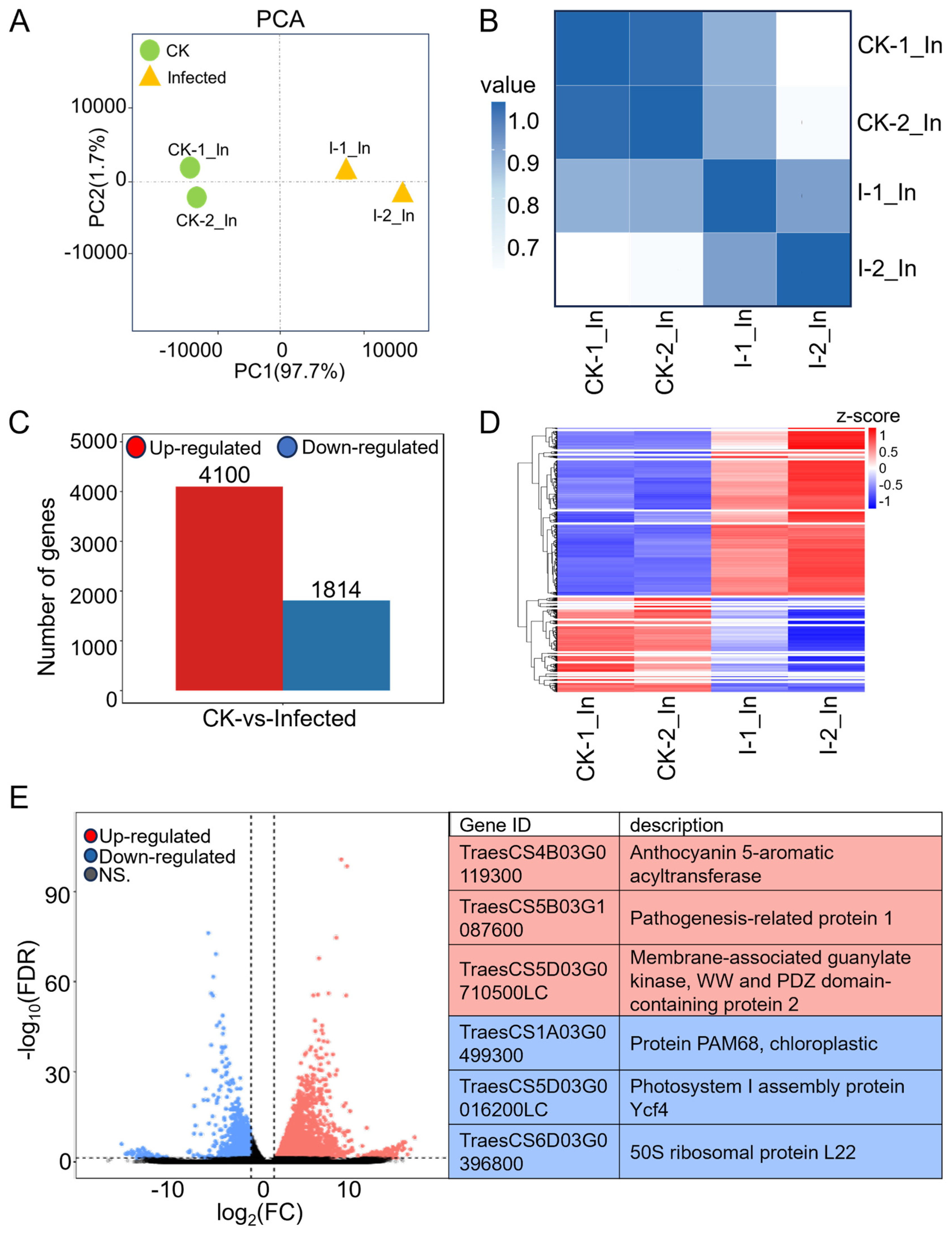
2.1. Transcriptome Profile of Wheat under Compatible Pst Interactions
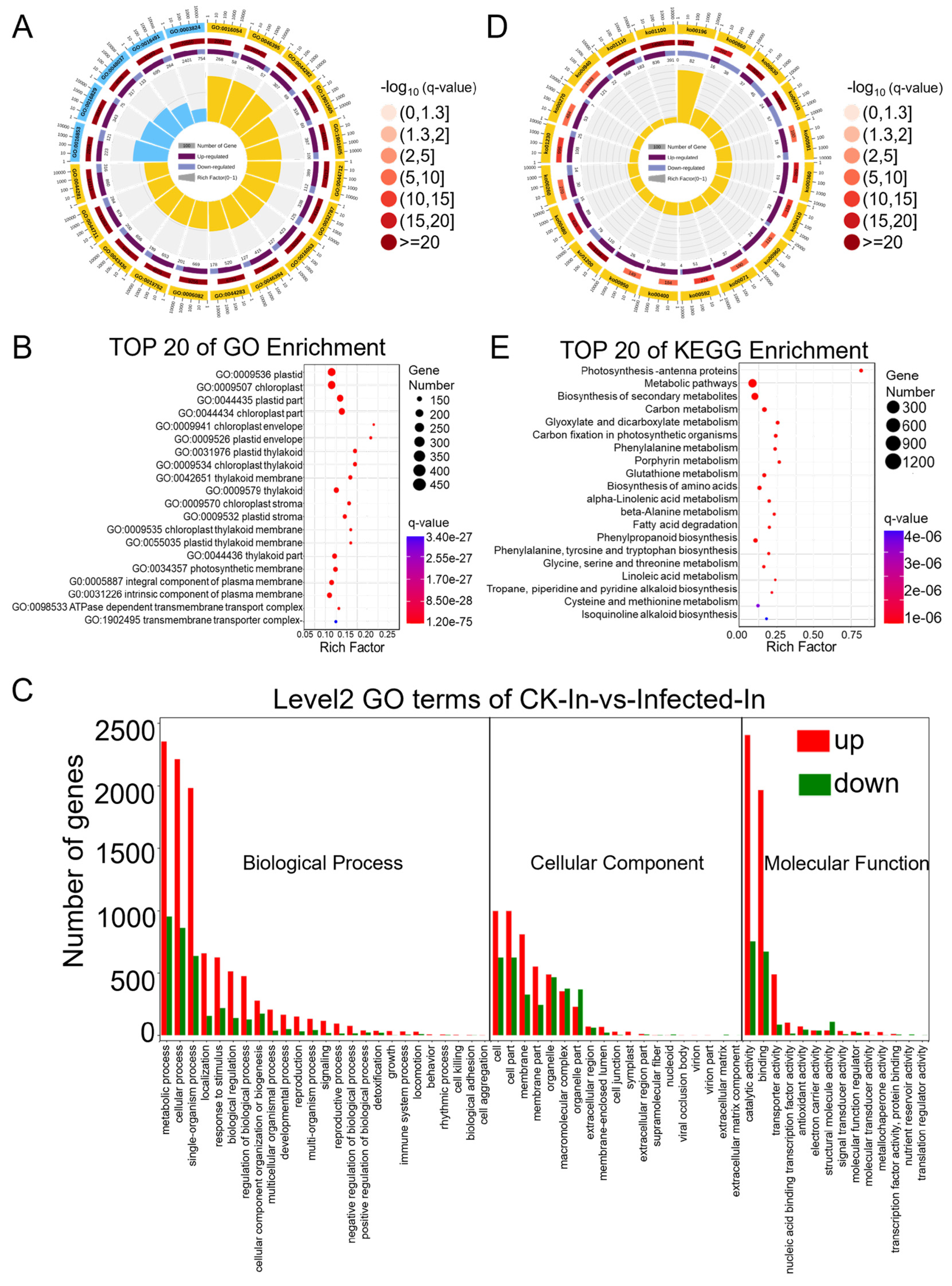
2.2. Most Differentially Expressed Genes (DEGs) in Wheat Were Induced by Pst Infection, while a Significant Decrease Was Observed in Genes Related to Photosynthesis
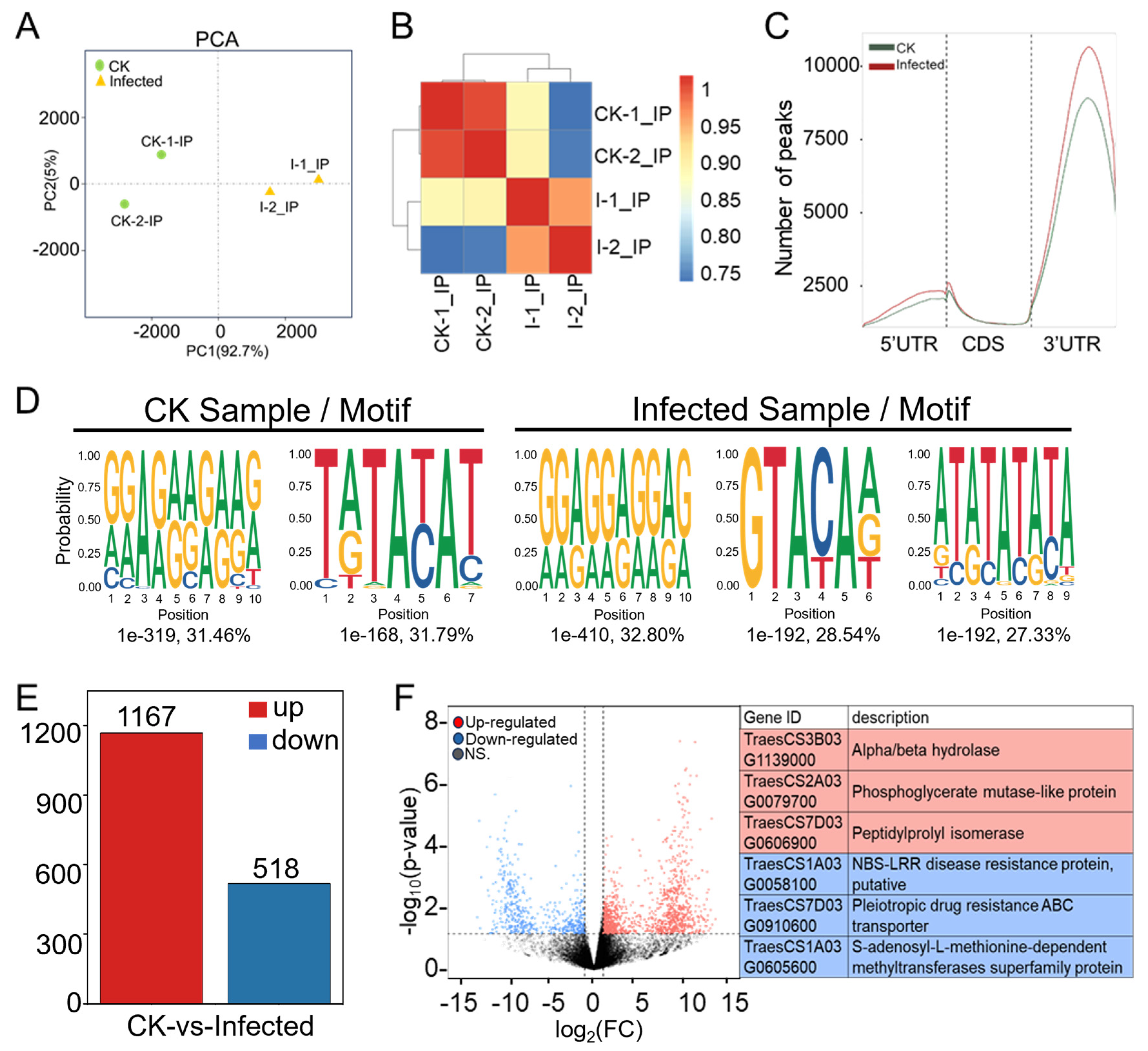
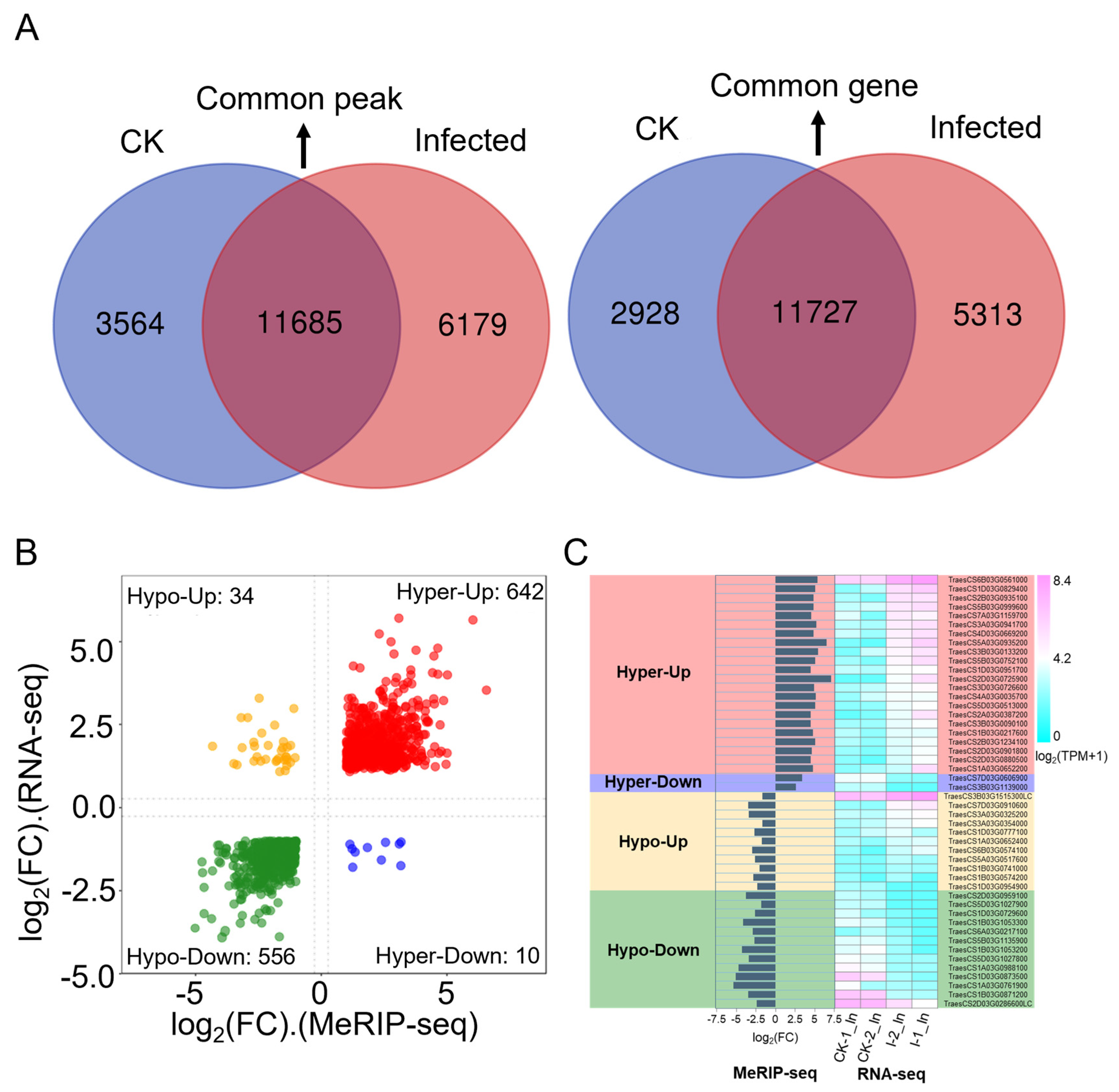
2.3. The Landscape of m6A Methylome in Pst-Infected Wheat Leaves
2.4. The Majority of Wheat Genes Associated with Differentially Methylated Peaks (DMPs) Showed Hyper-Methylation in Response to Pst
2.5. Association Analysis of DEGs and DMP-Related Genes in Pst-Infected Leaves in Comparison to CK
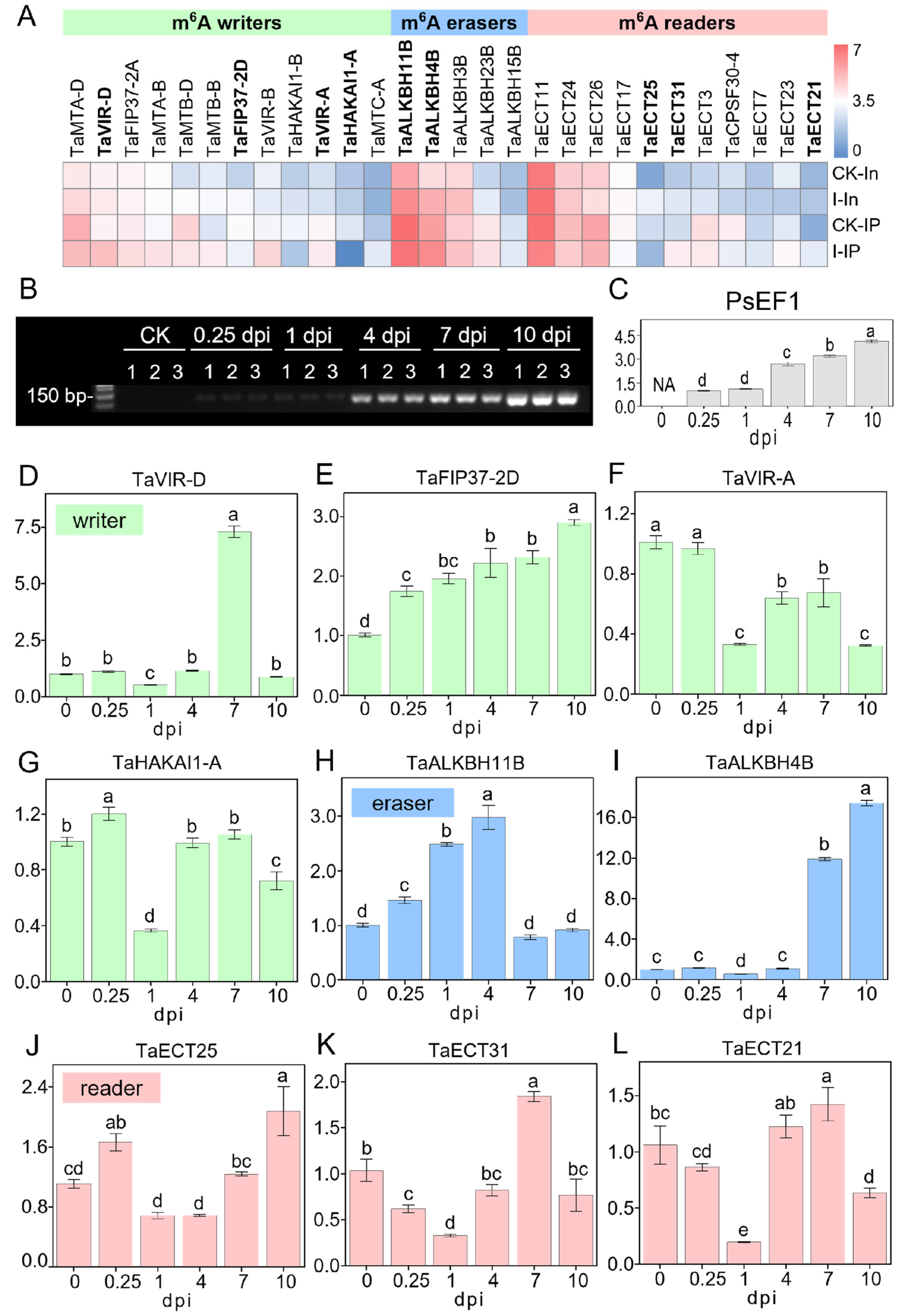
2.6. Divergent Expression Dynamics of m6A Regulators in Response to Pst Invasion
3. Discussion
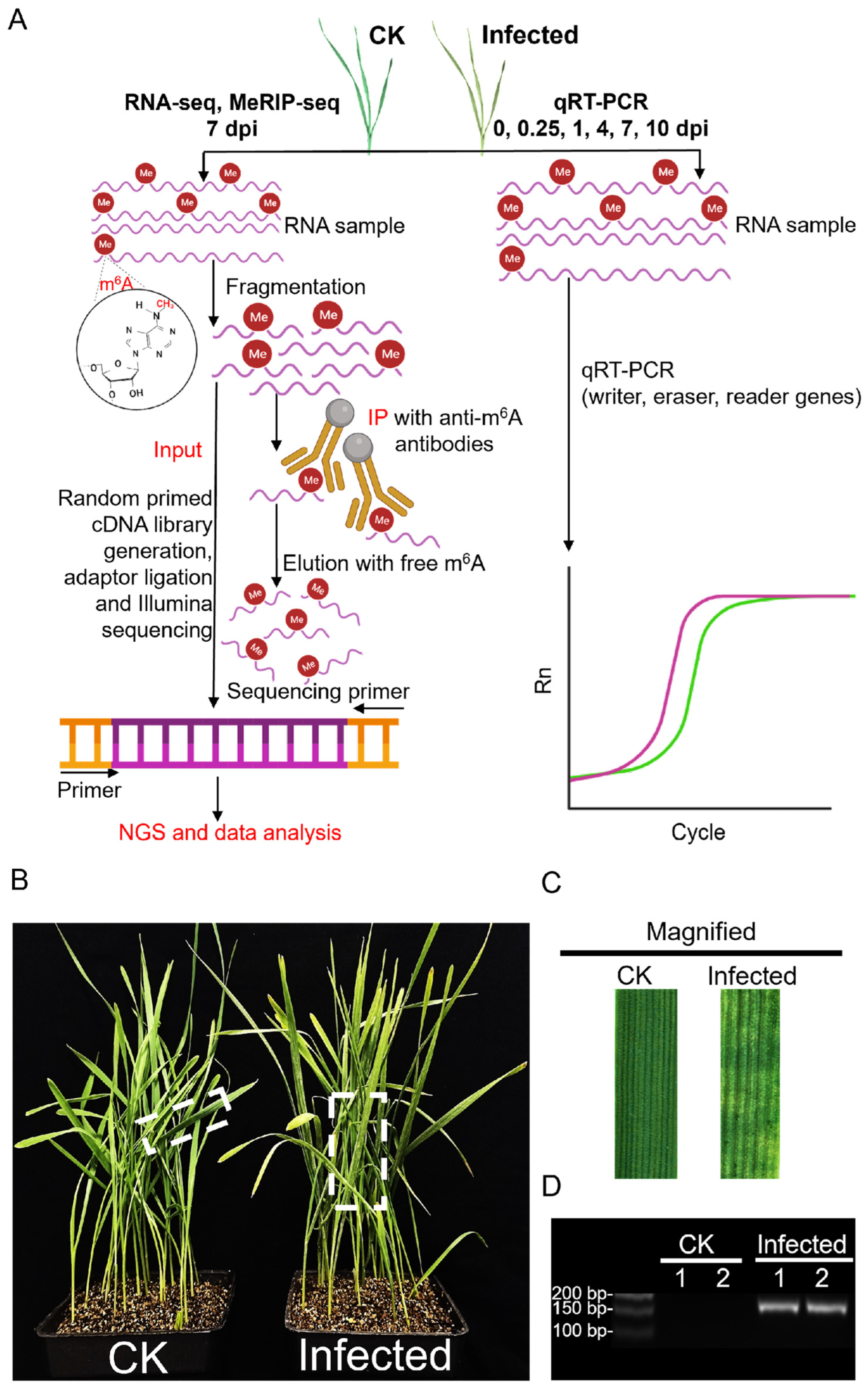
4. Materials and Methods
4.1. Plant Materials and Pst Inoculation
4.2. RNA Extraction and Library Construction for RNA-Seq and MeRIP-Seq
4.3. Data Analysis of Sequencing
4.4. sqRT-PCR and qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2021, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, L.; Li, X. Detection technologies for RNA modifications. Exp. Mol. Med. 2022, 54, 1601–1616. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Liu, Y.; Zhao, Y.; Yao, Y.; Wu, R.; Liu, Q.; Wang, Y.; Wang, X. A dynamic reversible RNA N6-methyladenosine modification: Current status and perspectives. J. Cell. Physiol. 2019, 234, 7948–7956. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Shi, T.; Zhang, Y.; He, C.; Zhang, Q.; Mo, Z.; Pan, W.; Nie, X. Genome-wide identification, expression and evolution analysis of m6A writers, readers and erasers in Aegilops_tauschii. Plants 2023, 12, 2747. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ma, J.; Li, P.; Wu, Y.; Yu, H. Recent advances in the plant epitranscriptome. Genome Biol. 2023, 24, 43. [Google Scholar] [CrossRef] [PubMed]
- Shinde, H.; Dudhate, A.; Kadam, U.S.; Hong, J.C. RNA methylation in plants: An overview. Front. Plant Sci. 2023, 14, 1132959. [Google Scholar] [CrossRef] [PubMed]
- Rajecka, V.; Skalicky, T.; Vanacova, S. The role of RNA adenosine demethylases in the control of gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liang, Z.; Wong, C.E.; Yu, H. Messenger RNA modifications in plants. Trends Plant Sci. 2019, 24, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 39, 1581–1588. [Google Scholar] [CrossRef]
- Bai, B. An update on principles of m6A targeting. Trends Plant Sci. 2022, 27, 224–226. [Google Scholar] [CrossRef]
- Ma, W.; Cui, S.; Lu, Z.; Yan, X.; Cai, L.; Lu, Y.; Cai, K.; Zhou, H.; Ma, R.; Zhou, S.; et al. YTH domain proteins play an essential role in rice growth and stress response. Plants 2022, 11, 2206. [Google Scholar] [CrossRef]
- Tang, J.; Chen, S.; Jia, G. Detection, regulation, and functions of RNA N6-methyladenosine modification in plants. Plant Commun. 2023, 4, 100546. [Google Scholar] [CrossRef]
- Wang, K.; Peng, J.; Yi, C. The m6A consensus motif provides a paradigm of epitranscriptomic studies. Biochemistry 2021, 60, 3410–3412. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; He, W.J.; Li, C.; Zhang, J.B.; Liao, Y.C.; Song, B.; Yang, P. Transcriptome-wide analyses of RNA m6A methylation in hexaploid wheat reveal its roles in mRNA translation regulation. Front. Plant Sci. 2022, 13, 917335. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Hernández, L.; Brodersen, P. Occurrence and functions of m6A and other covalent modifications in plant mRNA. Plant Physiol. 2019, 182, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, C.; Li, S.; Yuan, M.; Mu, W.; Yang, J.; Ma, Y.; Guan, C.; Ma, C. Changes in m6A RNA methylation are associated with male sterility in wolfberry. BMC Plant Biol. 2023, 23, 456. [Google Scholar] [CrossRef] [PubMed]
- Chuong, N.N.; Doan, P.P.T.; Wang, L.; Kim, J.H.; Kim, J. Current insights into m6A RNA methylation and its emerging role in plant circadian clock. Plants 2023, 12, 624. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157.e3. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Zhang, T.; Qi, Y.; Song, J.; Han, Z.; Ma, C. Evolution of the RNA N6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020, 182, 345–360. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Hu, J.; Manduzio, S.; Kang, H. Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Gao, G.; Tang, R.; Wang, W.; Wang, Y.; Tian, S.; Qin, G. m6A-mediated regulation of crop development and stress responses. Plant Biotechnol. J. 2022, 20, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Rudy, E.; Grabsztunowicz, M.; Arasimowicz-Jelonek, M.; Tanwar, U.K.; Maciorowska, J.; Sobieszczuk-Nowicka, E. N6-methyladenosine (m6A) RNA modification as a metabolic switch between plant cell survival and death in leaf senescence. Front. Plant Sci. 2023, 13, 1064131. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhang, F.; Qiao, X.; Zhao, Y.; Qiao, Q.; Huang, X.; Zhang, S. Multi-Omics analysis reveals the dynamic changes of RNA N6-Methyladenosine in pear (Pyrus bretschneideri) defense responses to Erwinia amylovora pathogen infection. Front. Microbiol. 2022, 12, 803512. [Google Scholar] [CrossRef]
- Rathod, V.; Hamid, R.; Tomar, R.S.; Patel, R.; Padhiyar, S.; Kheni, J.; Thirumalaisamy, P.P.; Munshi, N.S. Comparative RNA-Seq profiling of a resistant and susceptible peanut (Arachis hypogaea) genotypes in response to leaf rust infection caused by Puccinia arachidis. 3 Biotech. 2020, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, T.; Dai, X.; Yang, D.; Wu, Y.; Chen, H.; Zheng, Y.; Zhi, Q.; Wan, X.; Tan, X. Comparative transcriptome analysis revealed molecular mechanisms of peanut leaves responding to Ralstonia solanacearum and its type III secretion system mutant. Front. Microbiol. 2022, 13, 998817. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lei, Y.; Wan, L.; Yan, L.; Lv, J.; Dai, X.; Ren, X.; Guo, W.; Jiang, H.; Liao, B. Comparative transcript profiling of resistant and susceptible peanut post-harvest seeds in response to aflatoxin production by Aspergillus flavus. BMC Plant Biol. 2016, 16, 54. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, H.; Ren, J.; Dong, J.; Zhao, X.; Wang, X.; Wang, J.; Zhong, C.; Zhao, S.; Liu, X.; et al. Comparative Transcriptome-based mining and expression profiling of transcription factors related to cold tolerance in peanut. Int. J. Mol. Sci. 2020, 21, 1921. [Google Scholar] [CrossRef]
- Chen, Y.; Ren, X.; Zhou, X.; Huang, L.; Yan, L.; Lei, Y.; Liao, B.; Huang, J.; Huang, S.; Wei, W.; et al. Dynamics in the resistant and susceptible peanut (Arachis hypogaea L.) root transcriptome on infection with the Ralstonia solanacearum. BMC Genomics. 2014, 15, 1078. [Google Scholar] [CrossRef]
- Xiao, D.; Li, X.; Zhou, Y.Y.; Wei, L.; Keovongkod, C.; He, H.; Zhan, J.; Wang, A.Q.; He, L.F. Transcriptome analysis reveals significant difference in gene expression and pathways between two peanut cultivars under Al stress. Gene 2021, 781, 145535. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, C.; Yang, G.; Feng, T.; Gao, H.; Wang, X.; Chi, X.; Xu, Y.; Wu, Q.; Chen, D. Identification of chilling-responsive transcripts in peanut (Arachis hypogaea L.). Electron. J. Biotechnol. 2011, 14, 5. [Google Scholar]
- Kumar, K.R.R.; Kirti, P.B. Differential gene expression in Arachis diogoi upon interaction with peanut late leaf spot pathogen, Phaeoisariopsis personata and characterization of a pathogen induced cyclophilin. Plant Mol. Biol. 2011, 75, 497–513. [Google Scholar] [CrossRef]
- Kumar, D.; Kirti, P.B. The genus Arachis: An excellent resource for studies on differential gene expression for stress tolerance. Front. Plant Sci. 2023, 14, 1275854. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, Y.; Zhou, W.; Guo, Q.; Bai, B.; Shen, S.; Huang, G. Study on stripe rust (Puccinia striiformis) effect on grain filling and seed morphology building of special winter wheat germplasm Huixianhong. PLoS ONE 2019, 14, e0215066. [Google Scholar] [CrossRef]
- Zheng, W.; Huang, L.; Huang, J.; Wang, X.; Chen, X.; Zhao, J.; Guo, J.; Zhuang, H.; Qiu, C.; Liu, J.; et al. High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nat. Commun. 2013, 4, 2673. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lu, J.; Xing, J.; Xue, L.; Wu, Y.; Zhang, L. Characterization and functional analyses of wheat TaPR1 genes in response to stripe rust fungal infection. Sci. Rep. 2023, 13, 3362. [Google Scholar] [CrossRef]
- Meng, J.; Lu, Z.; Liu, H.; Zhang, L.; Zhang, S.; Chen, Y.; Rao, M.K.; Huang, Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 2014, 69, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Prall, W.; Ganguly, D.R.; Gregory, B.D. The covalent nucleotide modifications within plant mRNAs: What we know, how we find them, and what should be done in the future. Plant Cell 2023, 35, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Nie, X.; Yan, Z.; Weining, S. N6-methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol. J. 2019, 17, 1194–1208. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Feuillet, C.; Gallagher, D. The Wheat Genome, 1st ed.; Springer: Cham, Switzerland, 2023. [Google Scholar]
- Ozketen, A.C.; Andac-Ozketen, A.; Dagvadorj, B.; Demiralay, B.; Akkaya, M.S. In-depth secretome analysis of Puccinia striiformis f. sp. tritici in infected wheat uncovers effector functions. Biosci. Rep. 2020, 40, BSR20201188. [Google Scholar] [CrossRef]
- Wu, N.; Ozketen, A.C.; Cheng, Y.; Jiang, W.; Zhou, X.; Zhao, X.; Guan, Y.; Xiang, Z.; Akkaya, M.S. Puccinia striiformis f. sp. tritici effectors in wheat immune responses. Front. Plant Sci. 2022, 13, 1012216. [Google Scholar] [CrossRef]
- Wang, J.; Peng, Y.; Xu, Y.; Li, Z.; Zhan, G.; Kang, Z.; Zhao, J. Pan-genome analysis reveals a highly plastic genome and extensive secreted protein polymorphism in Puccinia striiformis f. sp. tritici. J. Genet. Genom. 2023. [Google Scholar] [CrossRef]
- Qu, Z.; Zhang, H.; Wang, Q.; Zhao, H.; Liu, X.; Fu, Y.; Lin, Y.; Xie, J.; Cheng, J.; Li, B.; et al. Exploring the symbiotic mechanism of a virus-mediated endophytic fungus in its host by dual unique molecular identifier-RNA sequencing. mSystems 2021, 6, e0081421. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.E.; Cui, J.M.; Su, Y.Q.; Yuan, S.; Yuan, M.; Zhang, H.Y. Influence of stripe rust infection on the photosynthetic characteristics and antioxidant system of susceptible and resistant wheat cultivars at the adult plant stage. Front. Plant Sci. 2015, 6, 779. [Google Scholar] [CrossRef]
- Wang, S.; Li, Q.P.; Wang, J.; Yan, Y.; Zhang, G.L.; Yan, Y.; Zhang, H.; Wu, J.; Chen, F.; Wang, X.; et al. YR36/WKS1-mediated phosphorylation of PsbO, an extrinsic member of photosystem II, inhibits photosynthesis and confers stripe rust resistance in wheat. Mol. Plant 2019, 12, 1639–1650. [Google Scholar] [CrossRef]
- Chang, C.Y.; Yang, S.X.; Zhang, M.Q.; Guo, Y.T.; Li, X.M.; Yan, Y.; Ding, C.H.; Niu, K.X.; Wang, M.L.; Li, Q.Q.; et al. Suppression of ZEAXANTHIN EPOXIDASE 1 restricts stripe rust growth in wheat. Plant Commun. 2023, 4, 100608. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tang, C.; Wang, X.; Sun, S.; Zhao, J.; Kang, Z.; Wang, X. An effector protein of the wheat stripe rust fungus targets chloroplasts and suppresses chloroplast function. Nat. Commun. 2019, 10, 5571. [Google Scholar] [CrossRef] [PubMed]
- Andac, A.; Ozketen, A.C.; Dagvadorj, B.; Akkaya, M.S. An effector of Puccinia striiformis f. sp. tritici targets chloroplasts with a novel and robust targeting signal. Eur. J. Plant Pathol. 2020, 157, 751–765. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Stark, R.; Brown, G. DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data; Bioconductor: Boston, MA, USA, 2011; 100p. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.C.; Wei, L.H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is an RNA N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Adams, T.M.; Olsson, T.S.G.; Ramírez-González, R.H.; Bryant, R.; Bryson, R.; Campos, P.E.; Fenwick, P.; Feuerhelm, D.; Hayes, C.; Henriksson, T.; et al. Rust expression browser: An open source database for simultaneous analysis of host and pathogen gene expression profiles with expVIP. BMC Genom. 2021, 22, 166. [Google Scholar] [CrossRef] [PubMed]
- Dobon, A.; Bunting, D.C.; Cabrera-Quio, L.E.; Uauy, C.; Saunders, D.G. The host-pathogen interaction between wheat and yellow rust induces temporally coordinated waves of gene expression. BMC Genom. 2016, 17, 380. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotations/Description/Gene Symbol | Gene ID | Regulation |
|---|---|---|
| UDP-glucose: glycoprotein glucosyltransferase (UGGT) | TraesCS1A03G0409400 TraesCS1D03G0377700 | Up |
| Lectin receptor kinase (LECRK91) | TraesCS1B03G1249300 TraesCS1D03G1014400 | Up |
| Receptor-like protein kinase, putative, expressed (CRK6) | TraesCS2B03G0583500 | Up |
| GRAM domain family protein (VAD1) | TraesCS2B03G0620400 | Up |
| Cyclic nucleotide-gated channel (CNGC4) | TraesCS3A03G0774300 | Up |
| Calreticulin (CRT3) | TraesCS3A03G0936700 | Up |
| Pathogenesis-related protein PR-4 (PR4A, PR4B) | TraesCS3A03G1217600 TraesCS3B03G1460900 TraesCS3D03G1160600 | Up |
| Glutamate decarboxylase (SPL) | TraesCS3B03G0229400 TraesCS3D03G0168800 | Up |
| MACPF domain protein (CAD1) | TraesCS3D03G0628000 | Up |
| Pheophorbide a oxygenase, chloroplastic (PAO) | TraesCS4A03G1017200 TraesCS4B03G0809700 | Up |
| Receptor protein kinase, putative (RLK7) | TraesCS5A03G0031900 | Up |
| Cysteine protease (CCP2) | TraesCS5B03G0553900 | Up |
| Aminotransferase-like protein (OAT) | TraesCS5D03G0861400 | Up |
| Pleiotropic drug resistance ABC transporter (ABCG48) | TraesCS5D03G0992800 | Up |
| E3 ubiquitin-protein ligase (ATL6) | TraesCS6A03G0290200 | Up |
| Alcohol dehydrogenase, putative (CAD2) | TraesCS6A03G0429300 TraesCS6B03G0520600 TraesCS6D03G0363400 | Up |
| Allene oxide cyclase (AOC) | TraesCS6B03G1030800 TraesCS6D03G0731500 | Up |
| BZIP transcription factor protein (BZIP50) | TraesCS7A03G0964400 | Up |
| MACPF domain-containing protein (MACPF) | TraesCS7B03G0282300 | Up |
| WRKY transcription factor (WRKY28) | TraesCS7D03G1174900 | Up |
| GTPase Der (FZL) | TraesCS1A03G0630100 | Down |
| Apolipoprotein D (CHL) | TraesCS2D03G1058400 | Down |
| NAD(P)H-quinone oxidoreductase subunit N (ndhN) | TraesCS3A03G0952700 TraesCS3B03G1089400 TraesCS3D03G0886900 | Down |
| Telomere-associated protein (RIF1) | TraesCS7A03G0315600 | Down |
| Annotations/Description/Gene Symbol | Gene ID | Regulation |
|---|---|---|
| Glycerol kinase (GLPK) | TraesCS2B03G1367900, TraesCS2D03G1154600 | Up |
| Cysteine proteinase (CCP1) | TraesCS2A03G0676600, TraesCS2D03G0630100, TraesCS5A03G0791200, TraesCS5B03G0823900, TraesCS5D03G0747200 | Up |
| Calmodulin, putative (CML27, CML37, CML4) | TraesCS1B03G1255000, TraesCS1B03G1255400, TraesCS5A03G0918000 | Up |
| Heat shock protein 90 (HSP81-1) | TraesCS7A03G0555500, TraesCS7D03G0537900 | Up |
| Cyclic nucleotide-gated channel (CNGC2) | TraesCS5D03G0900900 | Up |
| Chaperone protein htpG family protein (HSP90-5, HSP90) | TraesCS5B03G0646100, TraesCS5D03G0595900, TraesCS7A03G1288100, TraesCS7B03G1204400, TraesCS7D03G1221800 | Up |
| Calcium-dependent protein kinase (CPK15, CPK9, CPK10, CPK8, CPK4) | TraesCS1D03G0995500, TraesCS4A03G0728000, TraesCS5A03G1009700, TraesCS5B03G1055900, TraesCS5D03G0955400, TraesCS5B03G1159600, TraesCS5D03G1051900, TraesCS6A03G0203200, TraesCS6B03G0271200 | Up |
| WRKY transcription factor (WRKY24) | TraesCS1A03G0172500, TraesCS1A03G0743700, TraesCS1B03G0235500, TraesCS1B03G0848500, TraesCS1D03G0166900, TraesCS1D03G0711800 | Up |
| Kinase family protein (PTI13) | TraesCS2D03G0794500 | Up |
| Respiratory burst oxidase (RBOHA, RBOHC, RBOHE) | TraesCS3A03G0705100, TraesCS4B03G0848700, TraesCS5A03G1171300, TraesCS5D03G0694900, TraesCS5B03G0560500, TraesCS5D03G0519000, TraesCS6D03G0366700 | Up |
| Calcium-binding protein (CML19, CML27, CML36, CML5, CML21, CML8, CML25) | TraesCS3B03G1378700, TraesCS4B03G0485600, TraesCS5A03G0382400LC, TraesCS5B03G0150400, TraesCS5B03G0370400, TraesCS5D03G0789100, TraesCS7B03G1109900, TraesCS7B03G1175200, TraesCS7D03G1164400 | Up |
| Pathogenesis-related protein 1-1 (PRMS) | TraesCS5A03G0484700, TraesCS5A03G1037300, TraesCS5A03G1037400, TraesCS5B03G0483900, TraesCS5B03G1087600, TraesCS5D03G0980700, TraesCS5D03G0980800, TraesCS7D03G0362800, TraesCS7D03G0450000 | Up |
| RPM1-interacting protein 4 (RIN4) | TraesCS7A03G0593800 | Up |
| NBS-LRR disease resistance protein, putative, expressed (RPM1, RPP13L4, PIK-2, PIK6-NP) | TraesCS1A03G0059400, TraesCS1D03G0032300, TraesCS2D03G0030700, TraesCS2D03G1070500, TraesCS3D03G0966800, TraesCSU03G0241100, TraesCS1D03G0036500, TraesCS7B03G0033000, TraesCS7D03G0011300 | Up |
| Disease resistance protein (NBS-LRR class) (RPS2) | TraesCS2A03G0930000, TraesCS2B03G1021500, TraesCS2D03G0868500 | Up |
| 3-ketoacyl-CoA synthase (FDH, KCS20, CUT1, KCS11) | TraesCS4A03G0012800, TraesCS4A03G0046100, TraesCS4B03G0728500, TraesCSU03G0063000LC, TraesCS7D03G0827800 | Up |
| Mitogen-activated protein kinase (MPK5) | TraesCS4A03G0218300 | Up |
| Elongation factor Tu (TUFA) | TraesCS6B03G0694200, TraesCS6D03G0480200 | Down |
| Gene ID, Strand a | Description b | Pattern c | Peak Annotation d | Peak Start e | Peak End f | IP_ L2FC g | In_ L2FC h |
|---|---|---|---|---|---|---|---|
| TraesCS6B03G0561000, + | Cytochrome P450 family protein | Hyper-up | 5′, start, CDS | 284010639 | 284010814 | 5.01 | 1.64 |
| TraesCS1D03G0829400, − | Chaperone DnaJ | Hyper-up | stop, 3′ | 440701051 | 440701101 | 4.64 | 4.02 |
| TraesCS2B03G0935100, − | Malate synthase | Hyper-up | start, CDS | 526103000 | 526103125 | 4.48 | 4.57 |
| TraesCS5B03G0999600, + | Chitinase | Hyper-up | start, CDS | 583600714 | 583600764 | 4.50 | 3.12 |
| TraesCS7A03G1159700, + | Ferredoxin--NADP reductase 2 | Hyper-up | start, CDS | 676526476 | 676526576 | 4.23 | 2.61 |
| TraesCS3A03G0941700, − | Tropinone reductase-like protein | Hyper-up | 5′, start, CDS | 647714736 | 647714861 | 4.87 | 3.22 |
| TraesCS4D03G0669200, − | DUF1997 family protein | Hyper-up | start, CDS | 453670930 | 453670980 | 4.48 | 3.28 |
| TraesCS5A03G0935200, − | Cysteine protease, putative | Hyper-up | 5′, start, CDS | 588519570 | 588519733 | 6.03 | 5.66 |
| TraesCS3B03G0133200, − | D-Ala-D/L-Ala epimerase | Hyper-up | start, CDS | 39875470 | 39875545 | 5.02 | 3.99 |
| TraesCS5B03G0752100, − | Serine aminopeptidase S33 | Hyper-up | 5′, start, CDS | 479815861 | 479816011 | 4.64 | 4.80 |
| TraesCS1D03G0951700, + | GDP-mannose-3’,5’-epimerase | Hyper-up | 5′ | 474501754 | 474501829 | 4.19 | 3.12 |
| TraesCS2D03G0725900, + | Kinesin-like protein | Hyper-up | stop, 3′ | 407828185 | 407828373 | 6.57 | 3.53 |
| TraesCS3D03G0726600, + | 3-hydroxyacyl-CoA dehydrogenase | Hyper-up | stop, 3′ | 434958844 | 434958919 | 4.51 | 1.83 |
| TraesCS4A03G0035700, − | plant/protein | Hyper-up | start, CDS | 13580848 | 13580923 | 4.77 | 1.69 |
| TraesCS5D03G0513000, + | Jasmonate zim-domain protein | Hyper-up | start, CDS | 331598315 | 331598415 | 4.73 | 2.48 |
| TraesCS2A03G0387200, + | vacuolar sorting-associated protein | Hyper-up | CDS | 153236044 | 153236119 | 4.14 | 2.20 |
| TraesCS3B03G0090100, + | Nuclease S1 | Hyper-up | 5′, start, CDS | 24977420 | 24977620 | 4.13 | 2.69 |
| TraesCS1B03G0217600, + | AAA+ ATPase | Hyper-up | start, CDS | 75657827 | 75657977 | 4.38 | 1.63 |
| TraesCS2B03G1234100, − | Amine oxidase family protein | Hyper-up | 5′ | 695509349 | 695509524 | 4.68 | 1.63 |
| TraesCS2D03G0901800, − | SPX domain-containing protein | Hyper-up | stop, 3′ | 509963790 | 509963915 | 4.27 | 2.57 |
| TraesCS2D03G0880500, − | Aldehyde dehydrogenase | Hyper-up | stop, 3′ | 493975195 | 493975320 | 4.19 | 2.57 |
| TraesCS1A03G0652200, + | Pyruvate phosphate dikinase | Hyper-up | stop, 3′ | 446585467 | 446585567 | 4.43 | 4.06 |
| TraesCS7D03G0606900, + | Peptidylprolyl isomerase | Hyper-down | start, CDS | 254192941 | 254192991 | 3.17 | −1.75 |
| TraesCS3B03G1139000, + | Alpha/beta hydrolase | Hyper-down | CDS | 720605293 | 720605423 | 2.39 | −1.57 |
| TraesCS3B03G1515300LC, + | P-loop NTPase | Hypo-up | CDS, stop | 847160574 | 847160674 | −1.53 | 1.80 |
| TraesCS7D03G0910600, + | ABC transporter | Hypo-up | start, CDS | 502724870 | 502724945 | −3.19 | 2.70 |
| TraesCS3A03G0325200, + | GDSL esterase/lipase | Hypo-up | CDS, stop | 131460529 | 131460623 | −3.15 | 1.80 |
| TraesCS3A03G0354000, + | Cytochrome P450, putative | Hypo-up | CDS | 155007091 | 155007221 | −1.50 | 1.98 |
| TraesCS1D03G0777100, + | Cysteine protease family protein | Hypo-up | 5′, start | 421851652 | 421851802 | −2.47 | 3.29 |
| TraesCS1A03G0652400, − | Pyruvate phosphate dikinase | Hypo-up | start, CDS | 447084657 | 447084809 | −1.59 | 1.71 |
| TraesCS6B03G0574100, − | CASP-like protein | Hypo-up | 5′, start, CDS | 296054675 | 296054825 | −2.69 | 1.97 |
| TraesCS5A03G0517600, − | Homeobox protein, putative | Hypo-up | 5′ | 404317960 | 404318085 | −2.37 | 2.24 |
| TraesCS1B03G0741000, + | Pyruvate phosphate dikinase | Hypo-up | 5′, start | 471557711 | 471557786 | −1.85 | 2.48 |
| TraesCS1B03G0574200, − | Glutathione S-transferase | Hypo-up | 5′, start, CDS | 355708499 | 355708549 | −2.56 | 1.52 |
| TraesCS1D03G0954900, + | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 475689826 | 475689876 | −2.17 | −3.10 |
| TraesCS2D03G0959100, − | Cold-responsive proteinWCOR15 | Hypo-down | 5′, start, CDS | 540101549 | 540101599 | −3.47 | −2.90 |
| TraesCS5D03G1027900, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 511061211 | 511061286 | −1.67 | −3.89 |
| TraesCS1D03G0729600, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 405463983 | 405464083 | −2.37 | −3.47 |
| TraesCS1B03G1053300, + | Chlorophyll a/b binding protein | Hypo-down | start, CDS | 629357113 | 629357163 | −3.85 | −3.69 |
| TraesCS6A03G0217100, + | Chlorophyll a/b binding protein | Hypo-down | start, CDS | 64267362 | 64267412 | −2.64 | −3.37 |
| TraesCS5B03G1135900, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 638614728 | 638614778 | −2.47 | −3.18 |
| TraesCS1B03G1053200, + | Chlorophyll a/b binding protein | Hypo-down | start, CDS | 629262631 | 629262681 | −3.97 | −3.86 |
| TraesCS5D03G1027800, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 511058147 | 511058297 | −3.14 | −3.25 |
| TraesCS1A03G0988100, + | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 569868615 | 569868715 | −4.36 | −3.34 |
| TraesCS1D03G0873500, − | Chlorophyll a/b binding protein | Hypo-down | CDS | 454772861 | 454772911 | −4.66 | −3.19 |
| TraesCS1A03G0761900, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start, CDS | 500100318 | 500100462 | −5.01 | −3.65 |
| TraesCS1B03G0871200, − | Chlorophyll a/b binding protein | Hypo-down | 5′, start | 549194399 | 549194449 | −3.22 | −3.48 |
| TraesCS2D03G0286600LC, − | RING/U-box superfamily protein | Hypo-down | start, CDS | 82356920 | 82356989 | −2.24 | −3.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerav, E.N.; Wu, N.; Akkaya, M.S. Transcriptome-Wide N6-Methyladenosine (m6A) Methylation Analyses in a Compatible Wheat–Puccinia striiformis f. sp. tritici Interaction. Plants 2024, 13, 982. https://doi.org/10.3390/plants13070982
Cerav EN, Wu N, Akkaya MS. Transcriptome-Wide N6-Methyladenosine (m6A) Methylation Analyses in a Compatible Wheat–Puccinia striiformis f. sp. tritici Interaction. Plants. 2024; 13(7):982. https://doi.org/10.3390/plants13070982
Chicago/Turabian StyleCerav, Elif Naz, Nan Wu, and Mahinur S. Akkaya. 2024. "Transcriptome-Wide N6-Methyladenosine (m6A) Methylation Analyses in a Compatible Wheat–Puccinia striiformis f. sp. tritici Interaction" Plants 13, no. 7: 982. https://doi.org/10.3390/plants13070982
APA StyleCerav, E. N., Wu, N., & Akkaya, M. S. (2024). Transcriptome-Wide N6-Methyladenosine (m6A) Methylation Analyses in a Compatible Wheat–Puccinia striiformis f. sp. tritici Interaction. Plants, 13(7), 982. https://doi.org/10.3390/plants13070982