


Development of UHPLC-Q-Exactive Orbitrap/MS Technique for Determination of Proanthocyanidins (PAs) Monomer Composition Content in Persimmon


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Material
2.3. Instrumentation and Analytical Method
2.4. Extraction of PAs in Persimmon Fruit Flesh
2.5. Optimization of the Analytical Method
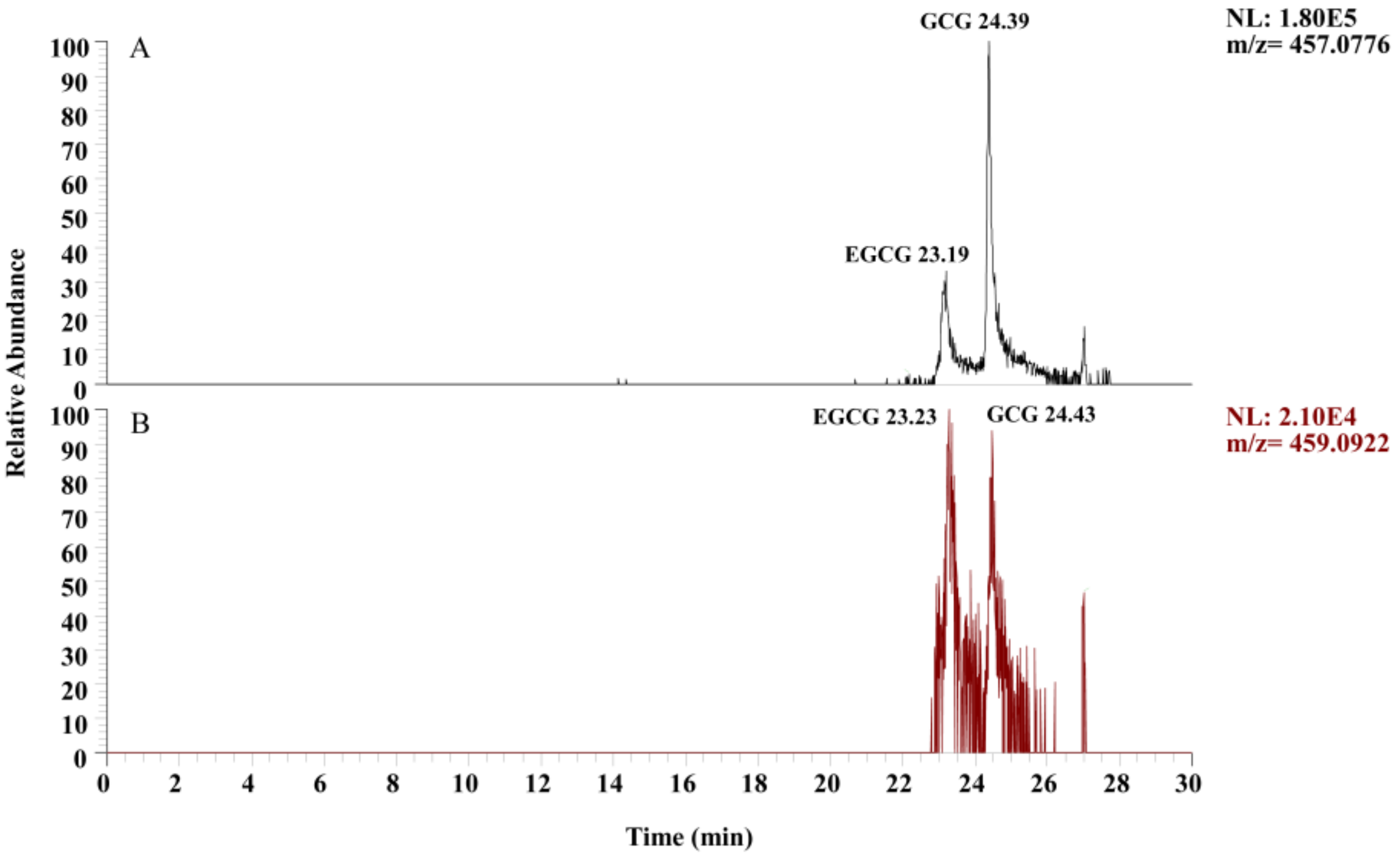
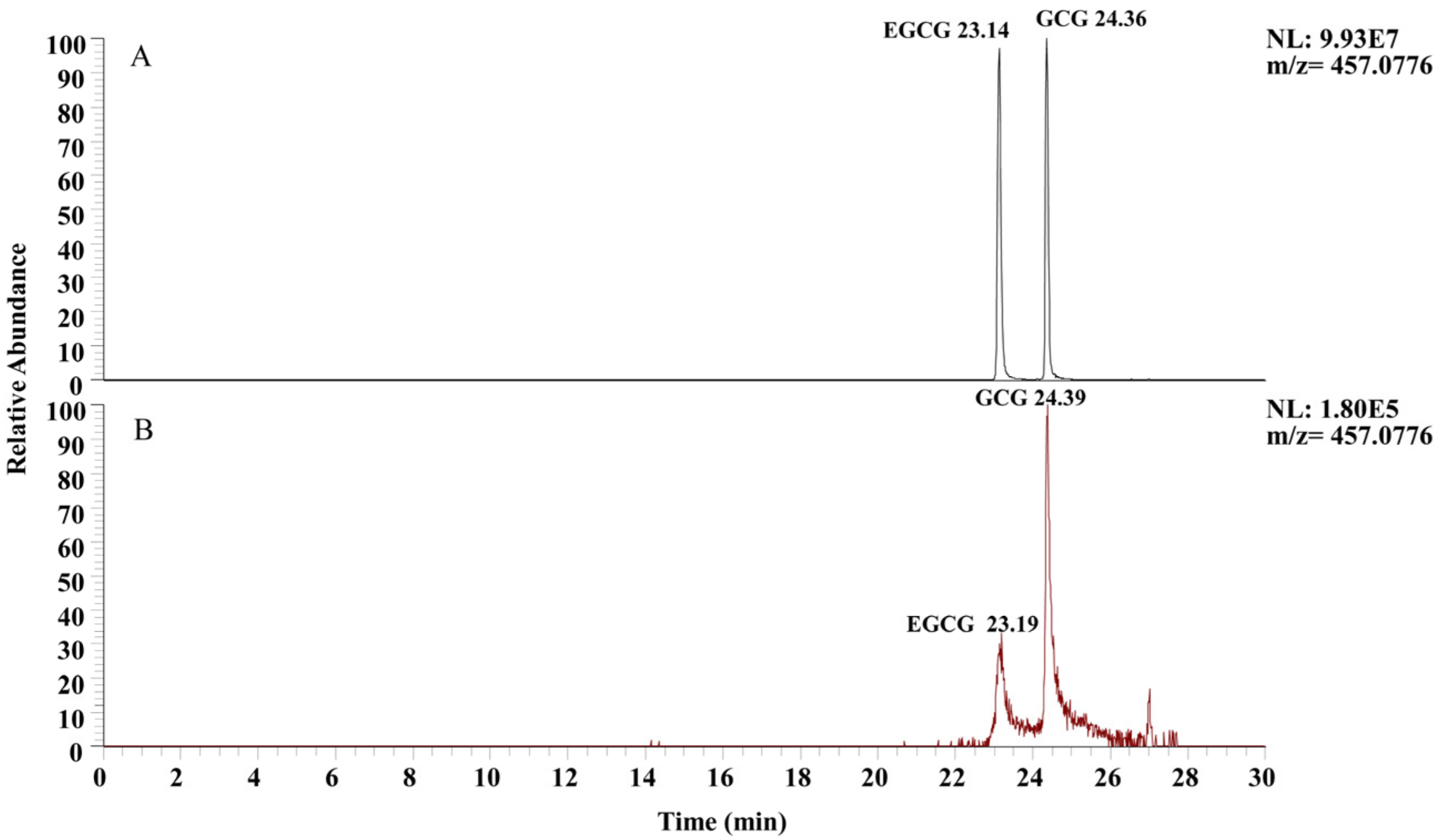
2.5.1. Selection of Ion Mode
2.5.2. Selection of Chromatographic Column
2.5.3. Selection of Mobile Phase
2.5.4. Selection of Mobile Phase Acid Content
2.5.5. Screening of Mobile Phase Flow Rate
2.6. Validation of Procedure Wide Suitability
2.7. Method Validation
2.7.1. Linearity
2.7.2. Test of Intra-Day and Inter-Day Precision
2.7.3. Test of Stability
2.7.4. Test of Recovery Rate
2.7.5. Limit of Detection and Quantification (LOD and LOQ)
2.8. Statistical Analysis
3. Results and Discussion
3.1. Selection of Ion Mode
3.2. Selection of Chromatographic Column
3.3. Selection of Mobile Phase
3.4. Selection of Mobile Phase Acid Content
3.5. Screening of Mobile Phase Flow Rate
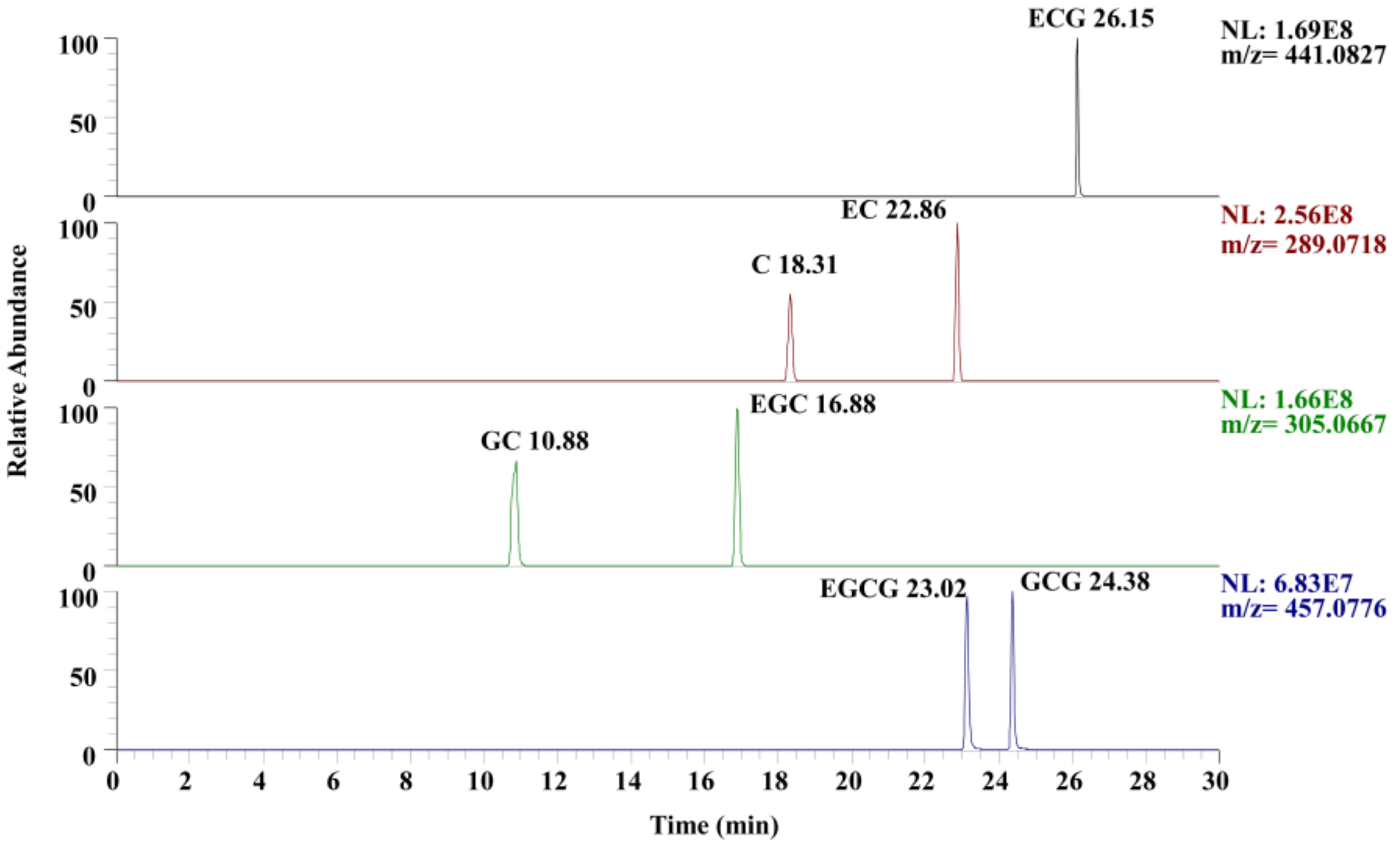
3.6. Validation of Procedure Wide Suitability
3.7. Linearity
3.8. Test of Intra-Day and Intra-Day Precision
3.9. Test of Stability
3.10. Test of Recovery
3.11. Limit of Detection and Quantification (LOD and LOQ)
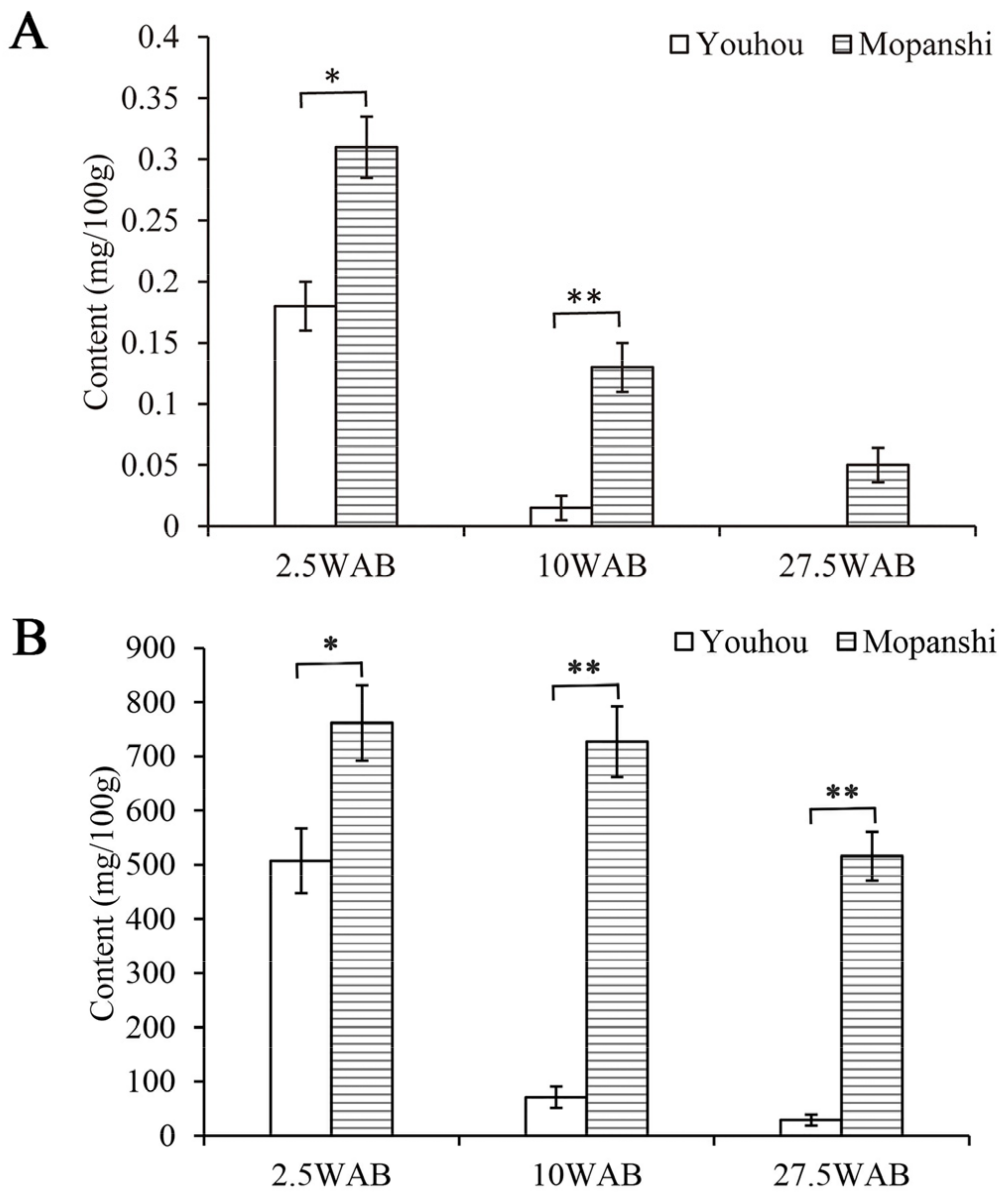
3.12. Procedure Applicability
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Taira, S.; Ono, M.; Matsumoto, N. Reduction of persimmon astringency by complex formation between pectin and tannins. Postharvest Biol. Biotechnol. 1997, 12, 265–271. [Google Scholar] [CrossRef]
- Aron, P.M.; Kennedy, J.A. Flavan-3-ols: Nature, occurrence and biological activity. Mol. Nutr. Food Res. 2008, 52, 79–104. [Google Scholar] [CrossRef]
- Watrelot, A.A.; Norton, E.L. Chemistry and Reactivity of Tannins in Vitis spp.: A Review. Molecules 2020, 25, 2110. [Google Scholar] [CrossRef]
- Hillis, W.E. Formation of condensed tannins in plants. Nature 1958, 182, 1371. [Google Scholar] [CrossRef]
- Matsuo, T.; Ito, S. The chemical structure of kaki-tannin from immature fruit of the persimmon (Diospyros kaki L.). Agric. Biol. Chem. 1978, 42, 1637–1643. [Google Scholar] [CrossRef]
- Gu, H.F.; Li, C.M.; Xu, Y.J.; Hu, W.F.; Chen, M.H.; Wan, Q.H. Structural features and antioxidant activity of tannin from persimmon pulp. Food Res. Int. 2008, 41, 208–217. [Google Scholar] [CrossRef]
- Li, C.M.; Leverence, R.; Trombley, J.D.; Xu, S.; Yang, J.; Tian, Y.; Reed, J.D.; Hagerman, A.E. High molecular weight persimmon (Diospyros kaki L.) proanthocyanidin: A highly galloylated, A-linked tannin with an unusual flavonol terminal unit, myricetin. J. Agric. Food Chem. 2010, 58, 9033–9042. [Google Scholar] [CrossRef]
- Dixon, R.A.; Xie, D.Y.; Sharma, S.B. Proanthocyanidins—A final frontier in flavonoid research? New Phytol. 2005, 165, 9–28. [Google Scholar] [CrossRef]
- Akagi, T.; Katayama-Ikegami, A.; Yonemori, K. Proanthocyanidin biosynthesis of persimmon (Diospyros kaki Thunb.) fruit. Sci. Hortic. 2011, 130, 373–380. [Google Scholar] [CrossRef]
- Xie, D.Y.; Sharma, S.B.; Paiva, N.L.; Ferreira, D.; Dixon, R.A. Role of anthocyanidin reductase, encoded by BANYULS in plant flavonoid biosynthesis. Science 2003, 299, 396–399. [Google Scholar] [CrossRef]
- Akagi, T.; Suzuki, Y.; Ikegami, A.; Kamitakahara, H.; Takano, T.; Nakatsubo, F.; Yonemori, K. Condensed tannin composition analysis in persimmon (Diospyros kaki Thunb.) fruit by acid catalysis in the presence of excess phloroglucinol. J. Jpn. Soc. Hortic. Sci. 2010, 79, 275–281. [Google Scholar] [CrossRef]
- Ancillotti, C.; Caprini, C.; Scordo, C.; Renai, L.; Giordani, E.; Orlandini, S.; Furlanetto, S.; Bubba, M.D. Phenolic compounds in Rojo Brillante and Kaki Tipo persimmons atcommercial harvest and in response to CO2 and ethylene treatments forastringency removal. LWT-Food Sci. Technol. 2019, 100, 99–105. [Google Scholar] [CrossRef]
- Chawla, G.; Chaudhary, K.K. A review of HPLC technique covering its pharmaceutical, environmental, forensic, clinical and other applications. Int. J. Pharm. Chem. Anal. 2019, 6, 27–39. [Google Scholar] [CrossRef]
- Yamamoto, R.; Genda, R.; Fukuhara, K.; Ogata, T.; Yonemori, K. Distribution of tannin cells in persimmon tree and the basic chemical composition of tannins in calluses derived from persimmon leaflets. Acta Hortic. 2022, 1338, 223–230. [Google Scholar] [CrossRef]
- Suzuki, T.; Someya, S.; Hu, F.Y.; Tanokura, M. Comparative study of catechin compositions in five Japanese persimmons (Diospyros kaki). Food Chem. 2005, 93, 149–152. [Google Scholar] [CrossRef]
- Veberic, R.; Jurhar, J.; Mikulic-Petkovsek, M.; Stampar, F.; Schmitzer, V. Comparative study of primary and secondary metabolites in 11 cultivars of persimmon fruit (Diospyros kaki L.). Food Chem. 2010, 119, 477–483. [Google Scholar] [CrossRef]
- Bordiga, M.; Travagliaa, F.; Giuffridab, D.; Mangravitic, D.; Riganod, F.; Mondello, L.; Arlorio, M.; Coïsson, J.D. Characterization of peel and pulp proanthocyanidins and carotenoids during ripening in persimmon ‘Kaki Tipo’ cv, cultivated in Italy. Food Res. Int. 2019, 120, 800–809. [Google Scholar] [CrossRef]
- Smith, D.M.; Wang, Z.G.; Kazi, A.; Li, L.H.; Chan, T.H.; Dou, D.P. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Mol. Med. 2002, 8, 382–392. [Google Scholar] [CrossRef]
- Benyahya, S.; Aouf, C.; Caillol, S.; Boutevin, B.; Pascault, J.P.; Fulcrand, H. Functionalized green tea tannins as phenolic prepolymers for bio-based epoxy resins. Ind. Crops Prod. 2014, 53, 296–307. [Google Scholar] [CrossRef]
- Zhao, L.S.; Li, F.M. UHPLC-MS strategies and applications for bioanalyses related to pharmacokinetics and drug metabolism. Trends Anal. Chem. 2014, 63, 170–179. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field orbitrap analyzer. Mol. Cell Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q exactive, a high performance benchtop quadrupole orbitrap mass spectrometer. Mol. Cell Proteom. 2011, 10, M111.011015. [Google Scholar] [CrossRef]
- Peel, G.J.; Dixon, R.A. Detection and quantification of engineered proanthocyanidins in transgenic plants. Nat. Prod. Commun. 2007, 2, 1009–1014. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Bioanalytical Method Validation. Guidance for Industry; U.S. Food and Drug Administration: Silver Spring, ML, USA, 2018.
- Kloos, D.P.; Lingeman, H.; Niessen, W.M.A.; Deelder, A.M.; Giera, M.; Mayborod, O.A. Evaluation of different column chemistries for fast urinary metabolic profiling. J. Chromatogr. B 2013, 927, 90–96. [Google Scholar] [CrossRef] [PubMed]
- New, L.S.; Chan, E.C.Y. Evaluation of BEH C18, BEH HILIC, and HSS T3 (C18) column chemistries for the UPLC-MS-MS analysis of glutathione, glutathione disulfide, and ophthalmic acid in mouse liver and human plasma. J. Chromatogr. Sci. 2008, 46, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A. Influence of mobile phase, source parametersand source type on electrospray ionization efficiency in negative ion mode. J. Mass. Spectrom. 2016, 51, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.J.; Carr, P.W. The surface tension of mixtures of methanol, acetonitrile, tetrahydrofuran, isopropanol, tertiary butanol and dimethyl-sulfoxide with water at 25 °C. J. Liq. Chromatogr. 1987, 10, 561–581. [Google Scholar] [CrossRef]
- Yun, K.Y.; Stringham, R.W. Effect of mobile phase acidic additives on enantioselectivity for phenylalanine analogs. J. Chromatogr. A 2001, 927, 47–52. [Google Scholar] [CrossRef]
- Barron, L.J.R.; Santa-María, G.; Díez Masa, J.C. Influence of bonded-phase column type, mobile phase composition, temperature and flow-rate in the analysis of triglycerides by reverse-phase high performance liquid chromatography. J. Liq. Chromatogr. 1987, 10, 3193–3212. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Volume Fraction of Flow Phase (%) | |
|---|---|---|
| A | B | |
| 0 | 98 | 2 |
| 5 | 95 | 5 |
| 15 | 90 | 10 |
| 20 | 85 | 15 |
| 24 | 70 | 30 |
| 25 | 0 | 100 |
| 27 | 0 | 100 |
| 27.1 | 98 | 2 |
| 30 | 98 | 2 |
| Compound | Regression Equation | R2 | Range of Linear μg/mL |
|---|---|---|---|
| EGCG | y = 831,335.87x + 100728.46 | 0.9991 | 0.1–100 |
| GCG | y = 2,272,957.25x − 26980.21 | 0.9998 | 0.01–10 |
| C | y = 2,412,557.34x − 307572.82 | 0.9993 | 0.05–20 |
| EC | y = 756,198.76x − 393787.58 | 0.9994 | 0.1–100 |
| ECG | y = 647,426.35x − 392818.31 | 0.9986 | 0.1–100 |
| GC | y = 2,821,093.03x − 276398.98 | 0.9984 | 0.05–20 |
| EGC | y = 520,917.04x − 269596.49 | 0.9992 | 0.1–100 |
| Compound | Spiked Amount/μg | Intra-Day Precision (RSD) | Inter-Day Precision (RSD) |
|---|---|---|---|
| EGCG | 200.00 | 1.60 | 1.75 |
| 50.00 | 1.59 | 1.83 | |
| 10.00 | 1.61 | 2.53 | |
| GCG | 200.00 | 1.32 | 1.72 |
| 50.00 | 1.36 | 2.23 | |
| 10.00 | 1.14 | 1.29 | |
| C | 200.00 | 1.83 | 1.45 |
| 50.00 | 2.11 | 2.92 | |
| 10.00 | 1.38 | 1.92 | |
| EC | 200.00 | 1.52 | 1.12 |
| 50.00 | 2.36 | 1.17 | |
| 10.00 | 1.65 | 2.07 | |
| GC | 200.00 | 2.12 | 2.71 |
| 50.00 | 1.39 | 1.34 | |
| 10.00 | 1.87 | 2.29 | |
| EGC | 200.00 | 1.32 | 1.12 |
| 50.00 | 1.18 | 1.23 | |
| 10.00 | 1.73 | 2.81 | |
| ECG | 200.00 | 2.02 | 1.14 |
| 50.00 | 1.86 | 1.03 | |
| 10.00 | 2.35 | 2.32 |
| Compound | Peak Area | Average | RSD/% | ||||
|---|---|---|---|---|---|---|---|
| 0 d | 2 d | 4 d | 8 d | 16 d | |||
| EGCG | 66,709,150.2 | 68,352,315.15 | 66,328,946.25 | 66,731,925.7 | 65,704,110.4 | 66,765,289.6 | 1.47 |
| GCG | 55,989.15 | 57,542.1 | 55,828.5 | 57,850.8 | 57,437.1 | 56,929.5 | 1.66 |
| C | 4,414,029.3 | 4,412,461.3 | 4,395,743 | 4,342,236.6 | 4,364,036.3 | 4,385,701.3 | 1.52 |
| EC | 131,509,817 | 136,223,418.3 | 133,189,066 | 131,890,391 | 131,018,021.7 | 132,766,142.8 | 1.98 |
| GC | 4,064,545 | 4,065,587.3 | 4,102,328.3 | 4,071,499.7 | 4,091,376.7 | 4,079,067.4 | 1.29 |
| EGC | 162,794,473 | 160,009,289.7 | 160,713,249.3 | 161,670,081.3 | 161,843,229.3 | 161,406,064.5 | 1.21 |
| ECG | 60,516,798.7 | 61,145,932.7 | 60,832,165 | 60,969,459.7 | 61,332,962 | 60,959,463.6 | 1.08 |
| Compound | Spiked Mean/mg | Recovery/% | RSD/% |
|---|---|---|---|
| EGCG | 5 | 96.89 | 1.25 |
| 2 | 95.25 | 1.66 | |
| 1 | 98.59 | 0.48 | |
| EC | 5 | 95.70 | 1.47 |
| 2 | 94.52 | 2.47 | |
| 1 | 96.32 | 3.70 | |
| EGC | 5 | 97.64 | 1.01 |
| 2 | 95.66 | 2.96 | |
| 1 | 96.42 | 0.61 | |
| ECG | 5 | 96.77 | 1.18 |
| 2 | 94.96 | 2.30 | |
| 1 | 96.09 | 2.32 | |
| GCG | 0.05 | 96.67 | 4.78 |
| 0.03 | 94.43 | 2.04 | |
| 0.02 | 97.45 | 1.08 | |
| C | 0.05 | 96.10 | 2.33 |
| 0.03 | 95.97 | 1.25 | |
| 0.02 | 94.67 | 1.99 | |
| GC | 0.05 | 95.58 | 1.05 |
| 0.03 | 95.30 | 2.35 | |
| 0.02 | 96.27 | 2.87 |
| Compound | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|
| EGCG | 0.00031 | 0.00103 |
| GCG | 0.00028 | 0.00094 |
| C | 0.00014 | 0.00046 |
| EC | 0.000077 | 0.00026 |
| ECG | 0.00017 | 0.00056 |
| GC | 0.00039 | 0.00129 |
| EGC | 0.000086 | 0.00029 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Ren, D.; Jin, R.; Chen, W.; Xu, L.; Guo, D.; Zhang, Q.; Luo, Z. Development of UHPLC-Q-Exactive Orbitrap/MS Technique for Determination of Proanthocyanidins (PAs) Monomer Composition Content in Persimmon. Plants 2024, 13, 1440. https://doi.org/10.3390/plants13111440
Zhao X, Ren D, Jin R, Chen W, Xu L, Guo D, Zhang Q, Luo Z. Development of UHPLC-Q-Exactive Orbitrap/MS Technique for Determination of Proanthocyanidins (PAs) Monomer Composition Content in Persimmon. Plants. 2024; 13(11):1440. https://doi.org/10.3390/plants13111440
Chicago/Turabian StyleZhao, Xianyang, Da Ren, Rui Jin, Wenxing Chen, Liqing Xu, Dayong Guo, Qinglin Zhang, and Zhengrong Luo. 2024. "Development of UHPLC-Q-Exactive Orbitrap/MS Technique for Determination of Proanthocyanidins (PAs) Monomer Composition Content in Persimmon" Plants 13, no. 11: 1440. https://doi.org/10.3390/plants13111440
APA StyleZhao, X., Ren, D., Jin, R., Chen, W., Xu, L., Guo, D., Zhang, Q., & Luo, Z. (2024). Development of UHPLC-Q-Exactive Orbitrap/MS Technique for Determination of Proanthocyanidins (PAs) Monomer Composition Content in Persimmon. Plants, 13(11), 1440. https://doi.org/10.3390/plants13111440