
The Chromatin Remodeling Factor BrCHR39 Targets DNA Methylation to Positively Regulate Apical Dominance in Brassica rapa

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
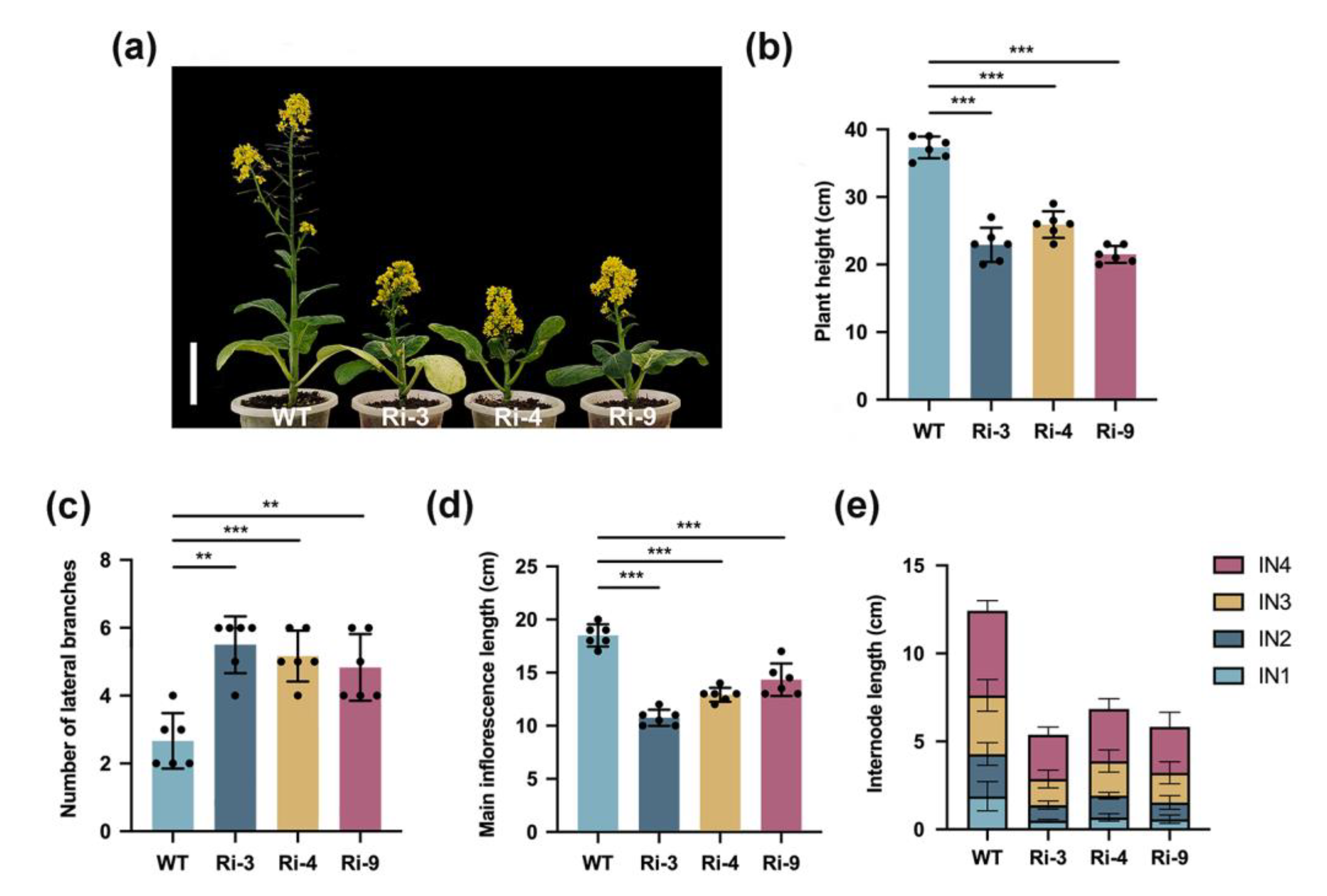
2.1. BrCHR39 Confers the Maintenance of Apical Dominance in Brassica rapa
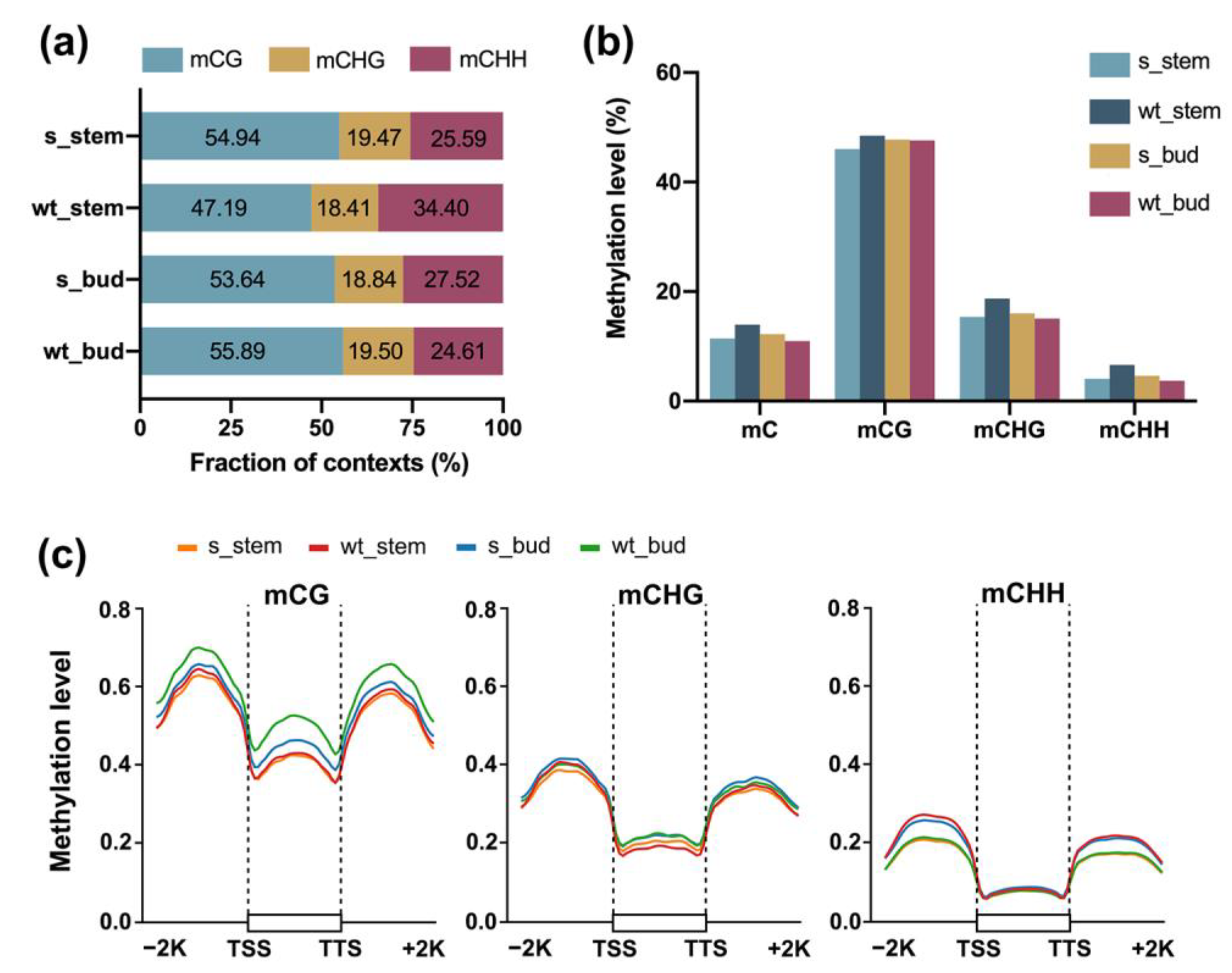
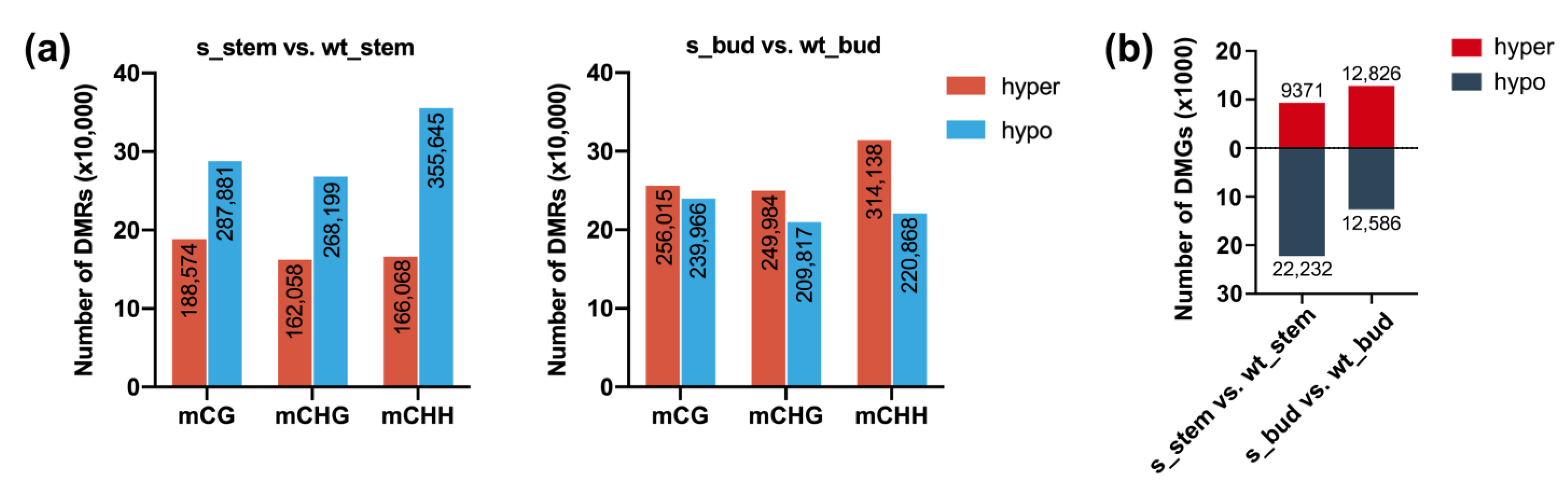
2.2. Profiles of Genome-Wide DNA Methylation in RNAi-BrCHR39 Plants and Wild-Type Plants
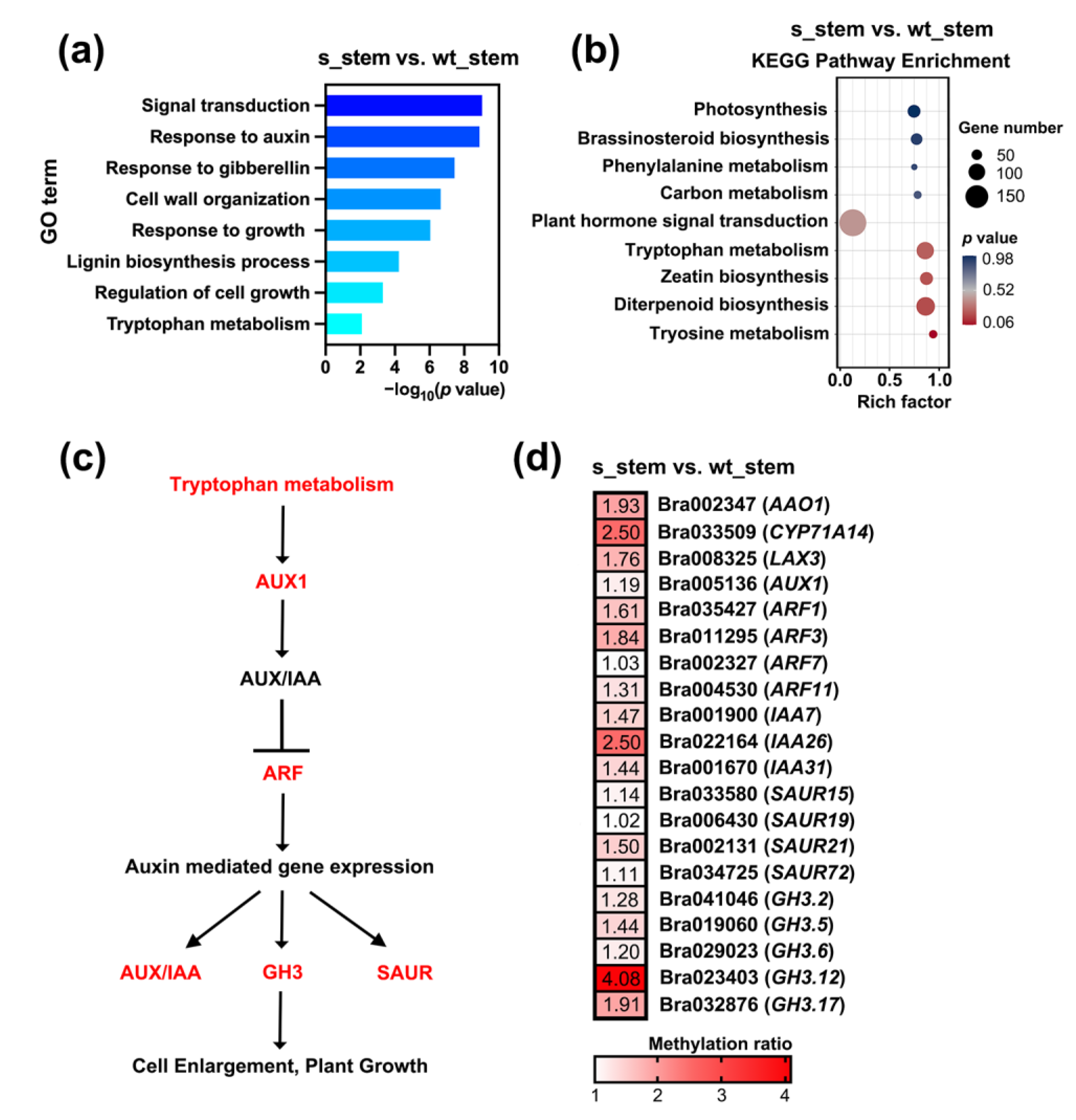
2.3. Hypermethylated Auxin-Related DMGs Resulted in Semi-Dwarfism in RNAi-BrCHR39 Plants
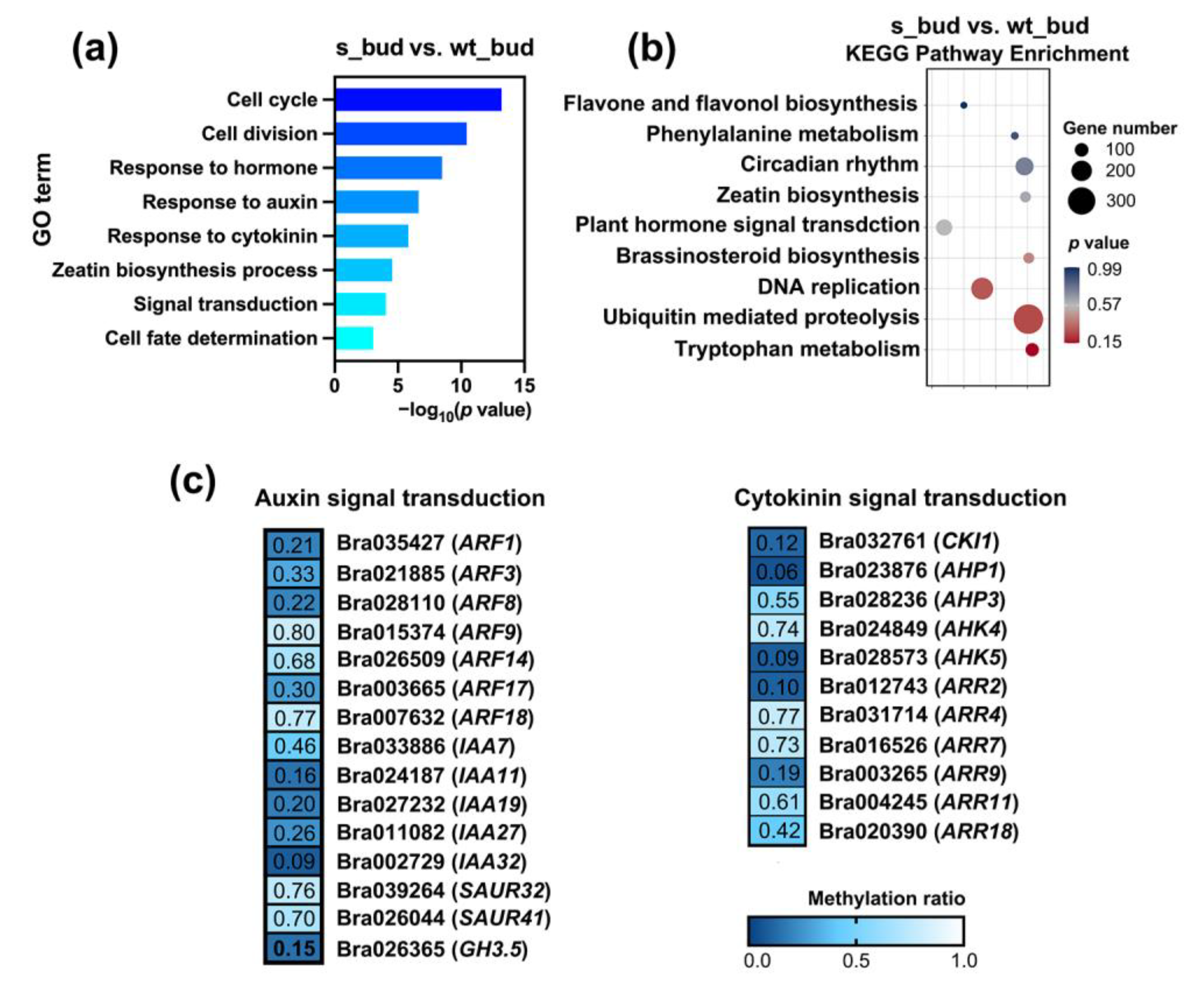
2.4. Hypomethylated Auxin and Cytokinin DMGs Promoted Bud Outgrowth in RNAi-BrCHR39 Plants
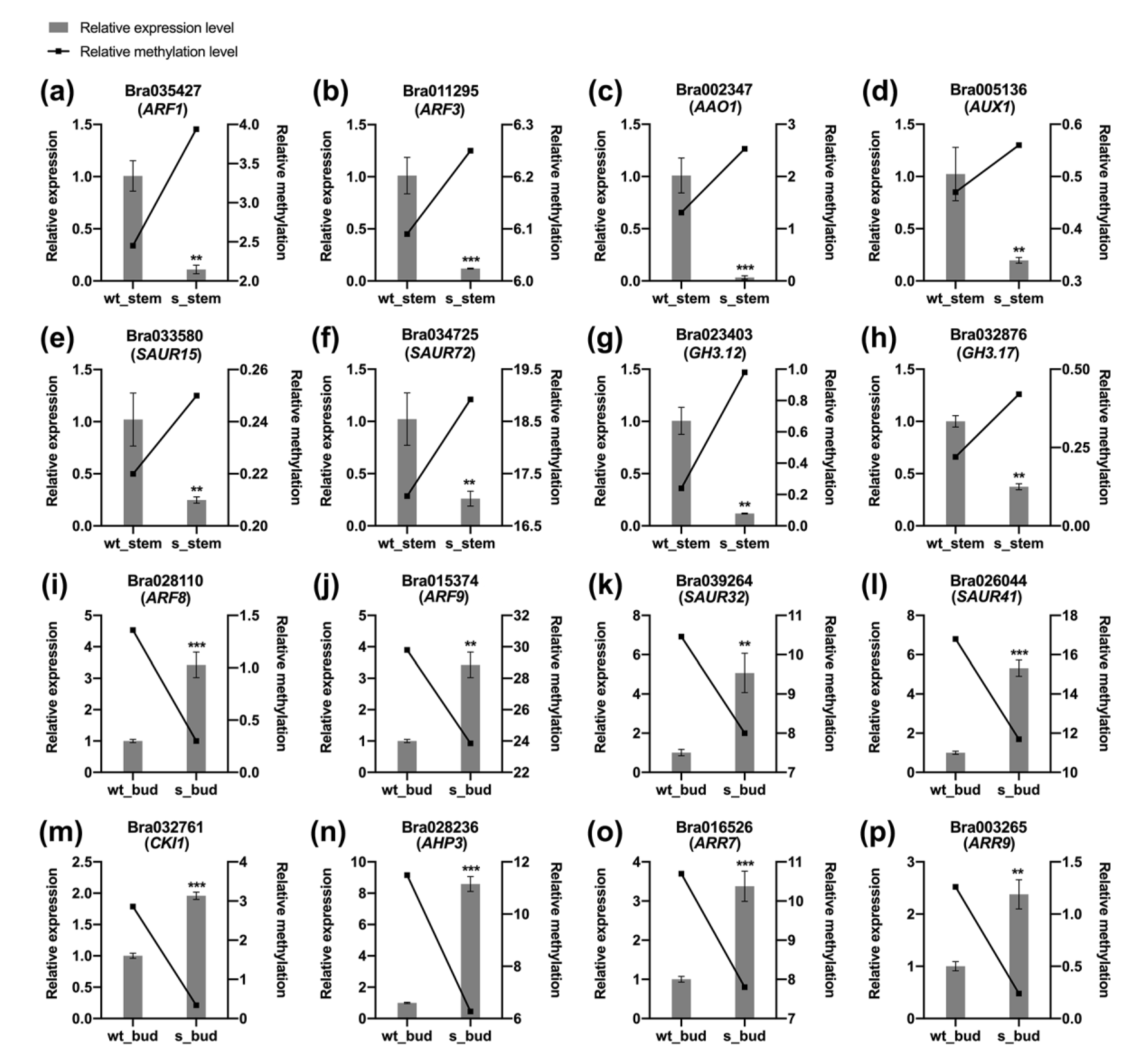
2.5. Expression Profiles of DMGs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Generation of Transgenic Plants
4.3. Whole-Genome Bisulfite Sequencing (WGBS) and Data Analysis
4.4. Differentially Methylated Region Analysis
4.5. GO and KEGG Enrichment Analysis of DMR-Related Genes
4.6. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Bba-Gene Regul. Mech. 2014, 1839, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Foroozani, M.; Holder, D.H.; Deal, R.B. Histone Variants in the Specialization of Plant Chromatin. Annu. Rev. Plant Biol. 2022, 73, 149–172. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Wang, L.; Qiao, H. Chromatin regulation in plant hormone and plant stress responses. Curr. Opin. Plant Biol. 2020, 57, 164–170. [Google Scholar] [CrossRef]
- Jarillo, J.A.; Pineiro, M.; Cubas, P.; Martinez-Zapater, J.M. Chromatin remodeling in plant development. Int. J. Dev. Biol. 2009, 53, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The Biology of Chromatin Remodeling Complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.Y.; He, X.J. Chromatin-remodeling complexes: Conserved and plant-specific subunits in Arabidopsis. J. Integr. Plant Biol. 2022, 64, 499–515. [Google Scholar] [CrossRef]
- Winston, F.; Carlson, M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992, 8, 387–391. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Lin, A.; Du, Y.; Xiao, W. Yeast chromatin remodeling complexes and their roles in transcription. Curr. Genet. 2020, 66, 657–670. [Google Scholar] [CrossRef]
- Ryan, D.P.; Owen-Hughes, T. Snf2-family proteins: Chromatin remodellers for any occasion. Curr. Opin. Chem. Biol. 2011, 15, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Pazin, M.J.; Kadonaga, J.T. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell 1997, 88, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.D. Mechanisms of ATP-dependent nucleosome sliding. Curr. Opin. Struct. Biol. 2010, 20, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Gerhold, C.B.; Lakomek, K.; Wollmann, P. Swi2/Snf2 remodelers: Hybrid views on hybrid molecular machines. Curr. Opin. Struct. Biol. 2012, 22, 225–233. [Google Scholar] [CrossRef]
- Flaus, A.; Martin, D.M.A.; Barton, G.J.; Owen-Hughes, T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef]
- Jones, P.L.; Veenstra, G.J.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef]
- Meyer, P. DNA methylation systems and targets in plants. FEBS Lett. 2011, 585, 2008–2015. [Google Scholar] [CrossRef]
- Lucibelli, F.; Valoroso, M.C.; Aceto, S. Plant DNA Methylation: An Epigenetic Mark in Development, Environmental Interactions, and Evolution. Int. J. Mol. Sci. 2022, 23, 8299. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Kovac, K.A. Plant DNA methyltransferases. Plant Mol. Biol. 2000, 43, 189–201. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Kass, S.U.; Pruss, D.; Wolffe, A.P. How does DNA methylation repress transcription? Trends Genet. 1997, 13, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Yang, R.; Zheng, Z.M.; Chen, Q.; Yang, L.; Huang, H.; Miki, D.; Wu, W.W.; Zeng, L.; Liu, J.; Zhou, J.X.; et al. The developmental regulator PKL is required to maintain correct DNA methylation patterns at RNA-directed DNA methylation loci. Genome Biol. 2017, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Li, J.; Cao, Y.; Huang, Y.; Li, W.; Zhu, H.; Zhao, Q.; Han, J.J.; Wu, Q.; Li, J.; et al. A role for LSH in facilitating DNA methylation by DNMT1 through enhancing UHRF1 chromatin association. Nucleic Acids Res. 2020, 48, 12116–12134. [Google Scholar] [CrossRef]
- Long, J.C.; Xia, A.A.; Liu, J.H.; Jing, J.L.; Wang, Y.Z.; Qi, C.Y.; He, Y. Decrease in DNA methylation 1 (DDM1) is required for the formation of (m) CHH islands in maize. J. Integr. Plant Biol. 2019, 61, 749–764. [Google Scholar] [CrossRef]
- Tan, F.; Lu, Y.; Jiang, W.; Wu, T.; Zhang, R.; Zhao, Y.; Zhou, D.X. DDM1 Represses Noncoding RNA Expression and RNA-Directed DNA Methylation in Heterochromatin. Plant Physiol. 2018, 177, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Coruh, C.; Xu, G.; Martins, L.M.; Bourbousse, C.; Lambolez, A.; Law, J.A. The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. Nat. Commun. 2022, 13, 244. [Google Scholar] [CrossRef]
- Brühl, J.; Trautwein, J.; Schäfer, A.; Linne, U.; Bouazoune, K. The DNA repair protein SHPRH is a nucleosome-stimulated ATPase and a nucleosome-E3 ubiquitin ligase. Epigenetics Chromatin 2019, 12, 52. [Google Scholar] [CrossRef]
- Kang, H.J.; Park, H.; Yoo, E.J.; Lee, J.H.; Choi, S.Y.; Lee-Kwon, W.; Lee, K.Y.; Hur, J.H.; Seo, J.K.; Ra, J.S.; et al. TonEBP Regulates PCNA Polyubiquitination in Response to DNA Damage through Interaction with SHPRH and USP1. iScience 2019, 19, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Ku, C.S.; Tsai, S.S.; Shiu, J.L.; Jiang, Y.Z.; Miriam, H.E.; Zhang, H.W.; Chen, Y.T.; Chiu, W.T.; Chang, S.B.; et al. PARP1 recruits DNA translocases to restrain DNA replication and facilitate DNA repair. PLoS Genet. 2022, 18, e1010545. [Google Scholar] [CrossRef]
- Tomi, N.S.; Davari, K.; Grotzky, D.; Loos, F.; Böttcher, K.; Frankenberger, S.; Jungnickel, B. Analysis of SHPRH functions in DNA repair and immunoglobulin diversification. DNA Repair. 2014, 24, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; An, J.; Park, Y.U.; Liaw, H.; Woodgate, R.; Park, J.H.; Myung, K. SHPRH regulates rRNA transcription by recognizing the histone code in an mTOR-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E3424–E3433. [Google Scholar] [CrossRef]
- Lee, D.; Park, J.H.; Kim, S.; Lee, S.G.; Myung, K. SHPRH as a new player in ribosomal RNA transcription and its potential role in homeostasis of ribosomal DNA repeats. Transcription 2018, 9, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. Chromatin Remodeling and Epigenetic Regulation in Plant DNA Damage Repair. Int. J. Mol. Sci. 2019, 20, 4093. [Google Scholar] [CrossRef]
- Leyser, O. The fall and rise of apical dominance-Commentary. Curr. Opin. Genet. Dev. 2005, 15, 468–471. [Google Scholar] [CrossRef]
- Tanaka, M.; Takei, K.; Kojima, M.; Sakakibara, H.; Mori, H. Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006, 45, 1028–1036. [Google Scholar] [CrossRef]
- Napoli, C.A.; Beveridge, C.A.; Snowden, K.C. Reevaluating concepts of apical dominance and the control of axillary bud outgrowth. Curr. Top. Dev. Biol. 1999, 44, 127–169. [Google Scholar]
- Barbier, F.F.; Dun, E.A.; Kerr, S.C.; Chabikwa, T.G.; Beveridge, C.A. An Update on the Signals Controlling Shoot Branching. Trends Plant Sci. 2019, 24, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.M.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef]
- Aichinger, E.; Villar, C.B.; Di Mambro, R.; Sabatini, S.; Kohler, C. The CHD3 chromatin remodeler PICKLE and polycomb group proteins antagonistically regulate meristem activity in the Arabidopsis root. Plant Cell 2011, 23, 1047–1060. [Google Scholar] [CrossRef]
- Ogas, J.; Cheng, J.C.; Sung, Z.R.; Somerville, C. Cellular differentiation regulated by gibberellin in the Arabidopsis thaliana pickle mutant. Science 1997, 277, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cao, L.; Rong, L.; An, Z.; Zhou, W.; Ma, J.; Shen, W.H.; Zhu, Y.; Dong, A. The chromatin-remodeling factor AtINO80 plays crucial roles in genome stability maintenance and in plant development. Plant J. 2015, 82, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Elserafy, M.; Abugable, A.A.; Atteya, R.; El-Khamisy, S.F. Rad5, HLTF, and SHPRH: A Fresh View of an Old Story. Trends Genet. 2018, 34, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Zeman, M.K.; Chen, J.Y.; Yee, M.C.; Cimprich, K.A. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 2011, 42, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdú, I.; Blastyák, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair. 2010, 9, 257–267. [Google Scholar] [CrossRef]
- Dhont, L.; Mascaux, C.; Belayew, A. The helicase-like transcription factor (HLTF) in cancer: Loss of function or oncomorphic conversion of a tumor suppressor? Cell Mol. Life Sci. 2016, 73, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Wulfridge, P.; Langmead, B.; Feinberg, A.P.; Hansen, K.D. Analyzing whole genome bisulfite sequencing data from highly divergent genotypes. Nucleic Acids Res. 2019, 47, e117. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Sun, M.; Zhang, T.; Li, H.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Yan, G.; et al. Global DNA methylation variations after short-term heat shock treatment in cultured microspores of Brassica napus cv. Topas. Sci. Rep. 2016, 6, 38401. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, X.; Wu, Z.; An, H.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Tu, J. Genome-Wide DNA Methylation Comparison between Brassica napus Genic Male Sterile Line and Restorer Line. Int. J. Mol. Sci. 2018, 19, 2689. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Picard, C.L.; Vong, B.; Feng, S.; Jacobsen, S.E. CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2125016118. [Google Scholar] [CrossRef]
- Schmitz, G.; Theres, K. Shoot and inflorescence branching. Curr. Opin. Plant Biol. 2005, 8, 506–511. [Google Scholar] [CrossRef]
- Kebrom, T.H. A Growing Stem Inhibits Bud Outgrowth-The Overlooked Theory of Apical Dominance. Front. Plant Sci. 2017, 8, 1874. [Google Scholar] [CrossRef]
- Karampelias, M.; Neyt, P.; De Groeve, S.; Aesaert, S.; Coussens, G.; Rolcik, J.; Bruno, L.; De Winne, N.; Van Minnebruggen, A.; Van Montagu, M.; et al. ROTUNDA3 function in plant development by phosphatase 2A-mediated regulation of auxin transporter recycling. Proc. Natl. Acad. Sci. USA 2016, 113, 2768–2773. [Google Scholar] [CrossRef]
- Balla, J.; Kalousek, P.; Reinohl, V.; Friml, J.; Prochazka, S. Competitive canalization of PIN-dependent auxin flow from axillary buds controls pea bud outgrowth. Plant J. 2011, 65, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Leyser, O. Regulation of shoot branching by auxin. Trends Plant Sci. 2003, 8, 541–545. [Google Scholar] [CrossRef]
- Schaller, G.E.; Bishopp, A.; Kieber, J.J. The yin-yang of hormones: Cytokinin and auxin interactions in plant development. Plant Cell 2015, 27, 44–63. [Google Scholar] [CrossRef] [PubMed]
- Zurcher, E.; Muller, B. Cytokinin Synthesis, Signaling, and Function--Advances and New Insights. Int. Rev. Cell Mol. Biol. 2016, 324, 1–38. [Google Scholar] [CrossRef]
- Müller, D.; Waldie, T.; Miyawaki, K.; To, J.P.; Melnyk, C.W.; Kieber, J.J.; Kakimoto, T.; Leyser, O. Cytokinin is required for escape but not release from auxin mediated apical dominance. Plant J. 2015, 82, 874–886. [Google Scholar] [CrossRef]
- Rubio-Moraga, A.; Ahrazem, O.; Pérez-Clemente, R.M.; Gómez-Cadenas, A.; Yoneyama, K.; López-Ráez, J.A.; Molina, R.V.; Gómez-Gómez, L. Apical dominance in saffron and the involvement of the branching enzymes CCD7 and CCD8 in the control of bud sprouting. BMC Plant Biol. 2014, 14, 171. [Google Scholar] [CrossRef]
- Fray, R.; Zhong, S. Genome-wide DNA methylation in tomato. Appl. Plant Genom. Biotechnol. 2015, 179–193. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- He, Y.; Li, Z. Epigenetic Environmental Memories in Plants: Establishment, Maintenance, and Reprogramming. Trends Genet. 2018, 34, 856–866. [Google Scholar] [CrossRef]
- Brukhin, V.; Albertini, E. Epigenetic Modifications in Plant Development and Reproduction. Epigenomes 2021, 5, 25. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Zhang, X.R.; Henriques, R.; Lin, S.S.; Niu, Q.W.; Chua, N.H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 2006, 1, 641–646. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Mapped Reads | Mapping Rate (%) | Bisulfite Conversion Rate (%) |
|---|---|---|---|---|---|
| s_stem | 67,906,410 | 67,853,544 | 32,396,178 | 47.74 | 99.75 |
| s_stem | 67,906,410 | 67,853,544 | 32,396,178 | 47.74 | 99.75 |
| wt_stem | 63,995,464 | 63,939,594 | 30,980,118 | 48.45 | 99.71 |
| s_bud | 67,422,054 | 67,365,438 | 32,592,740 | 48.38 | 99.73 |
| wt_bud | 59,469,540 | 59,420,634 | 28,819,630 | 48.50 | 99.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Xie, Z.; Chu, Z.; Ding, Y.; Shi, G.; Chen, W.; Wei, X.; Yuan, Y.; Wei, F.; Tian, B. The Chromatin Remodeling Factor BrCHR39 Targets DNA Methylation to Positively Regulate Apical Dominance in Brassica rapa. Plants 2023, 12, 1384. https://doi.org/10.3390/plants12061384
Zhu W, Xie Z, Chu Z, Ding Y, Shi G, Chen W, Wei X, Yuan Y, Wei F, Tian B. The Chromatin Remodeling Factor BrCHR39 Targets DNA Methylation to Positively Regulate Apical Dominance in Brassica rapa. Plants. 2023; 12(6):1384. https://doi.org/10.3390/plants12061384
Chicago/Turabian StyleZhu, Wei, Zhengqing Xie, Zhenni Chu, Yakun Ding, Gongyao Shi, Weiwei Chen, Xiaochun Wei, Yuxiang Yuan, Fang Wei, and Baoming Tian. 2023. "The Chromatin Remodeling Factor BrCHR39 Targets DNA Methylation to Positively Regulate Apical Dominance in Brassica rapa" Plants 12, no. 6: 1384. https://doi.org/10.3390/plants12061384
APA StyleZhu, W., Xie, Z., Chu, Z., Ding, Y., Shi, G., Chen, W., Wei, X., Yuan, Y., Wei, F., & Tian, B. (2023). The Chromatin Remodeling Factor BrCHR39 Targets DNA Methylation to Positively Regulate Apical Dominance in Brassica rapa. Plants, 12(6), 1384. https://doi.org/10.3390/plants12061384