
Genome-Wide Association Studies of Biluochun Tea Plant Populations in Dongting Mountain and Comprehensive Identification of Candidate Genes Associated with Core Agronomic Traits by Four Analysis Models

 and
and
Abstract
:1. Introduction
2. Result
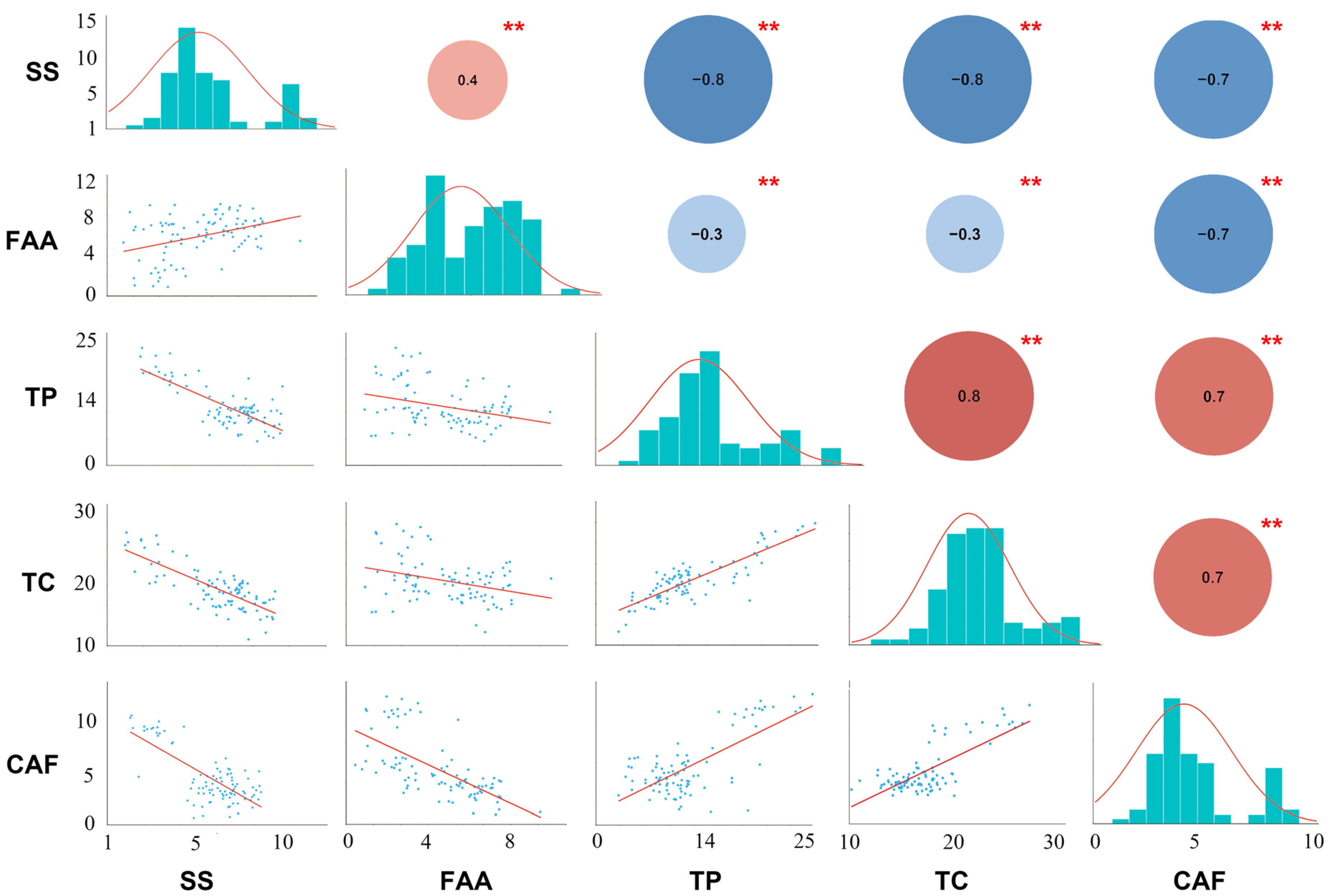
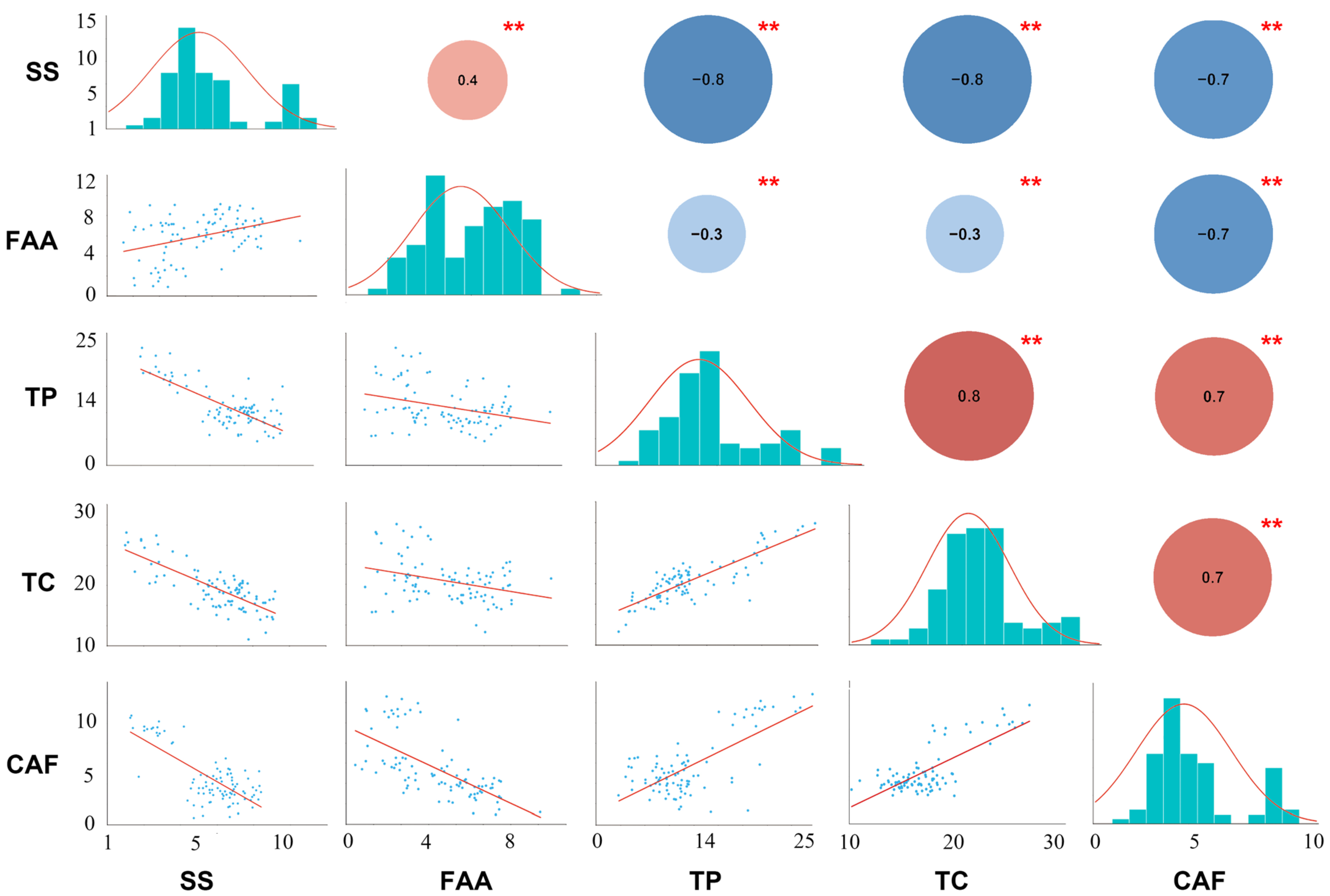
2.1. Metabolic Variation
2.2. Phenotypic Variation
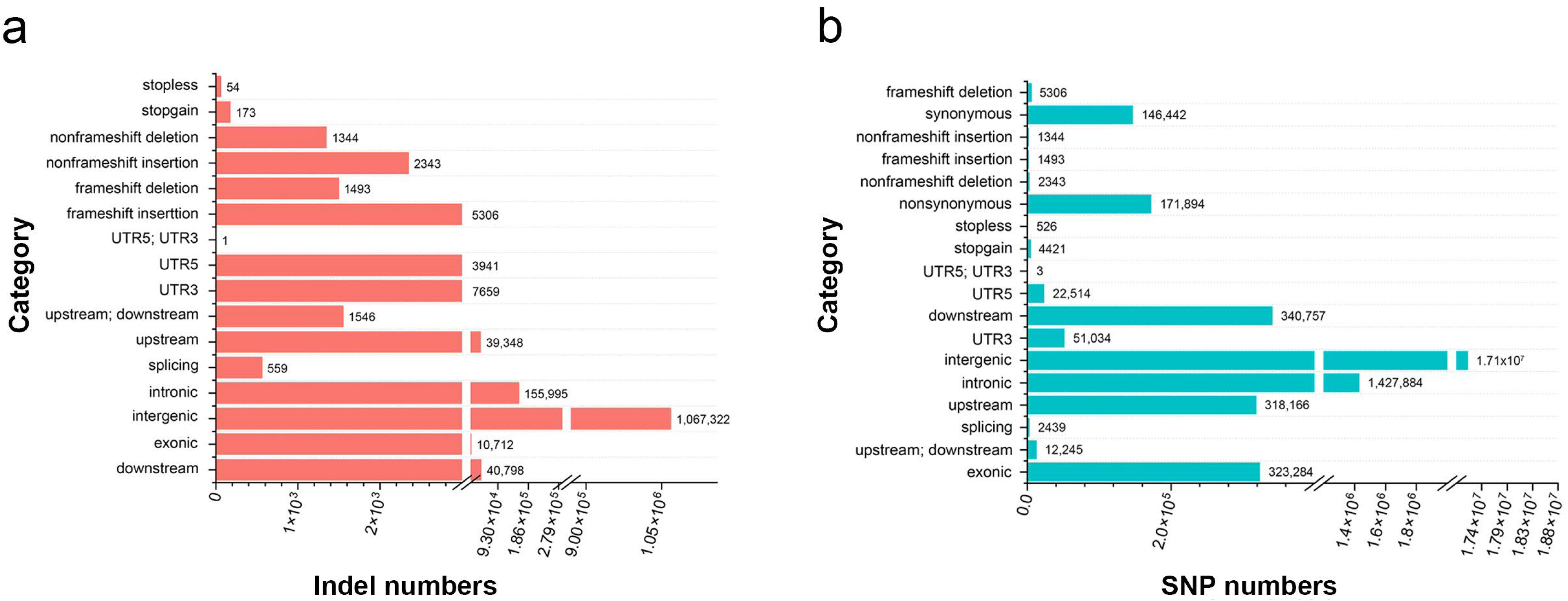
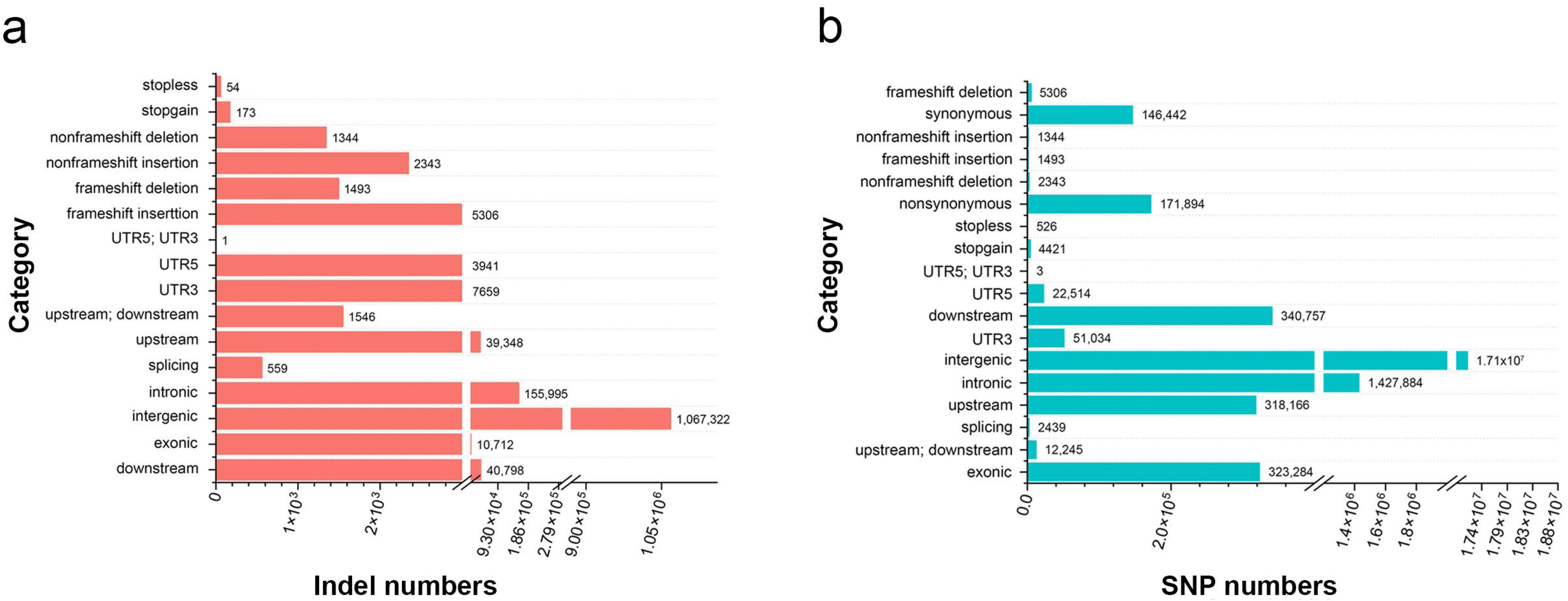
2.3. Sequencing and Variant Discovery
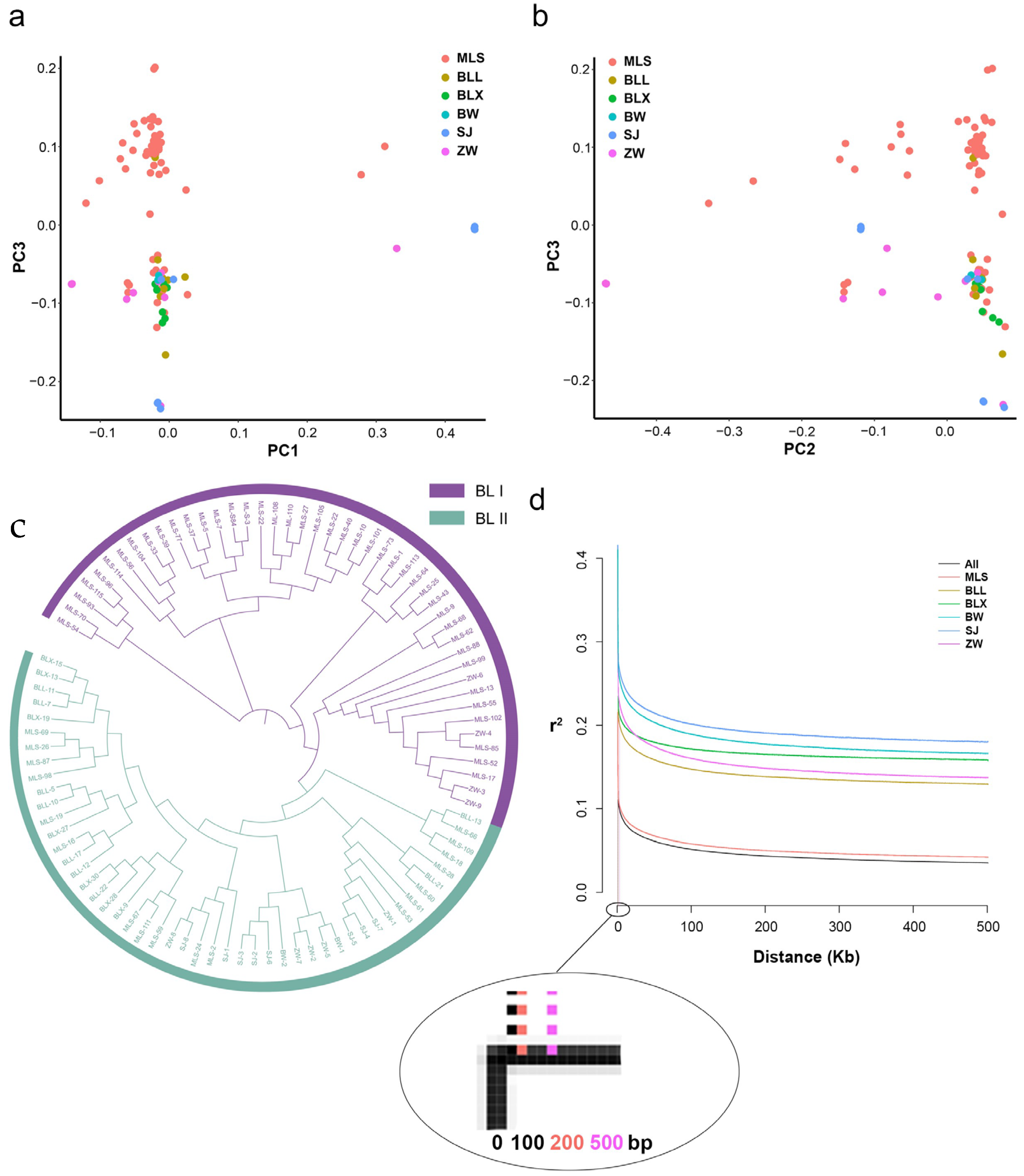
2.4. Phylogenetic Analysis and Population Structure of Biluochun Tea Plant Populations
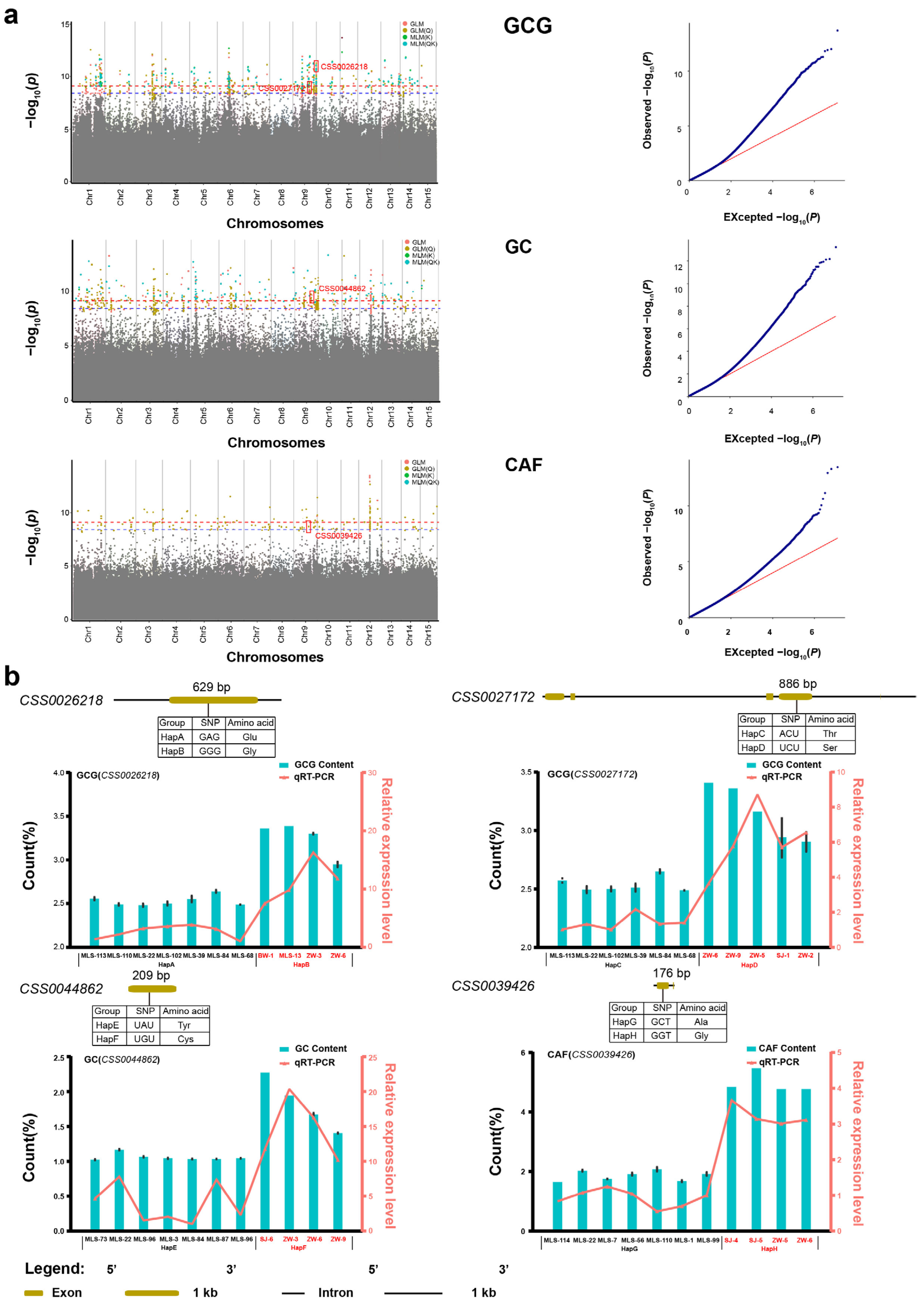
2.5. Genome-Wide Association Studies
2.6. Prediction of Potential Candidate Genes
3. Discussion
3.1. Population Structure and Genetic Evolution Analysis
3.2. Dissecting Four Candidate Genes of Quality-Related Traits
4. Materials and Methods
4.1. Plant Materials
4.2. Data Analysis
4.3. Sequencing, Mapping, and SNP Calling
4.4. Phylogenetic Tree and Population Genetics Analysis
4.5. Linkage Disequilibrium Analysis
4.6. Genome-Wide Association Study
4.7. Identification of Putative Genes
4.8. Expression Pattern Analysis
4.9. Determination of Each Quality Index of Tea
4.9.1. Chemicals
4.9.2. Determination of Chlorophylls
4.9.3. Determination of Free Amino Acids
4.9.4. Determination of Soluble Sugar
4.9.5. Determination of Tea Polyphenols
4.9.6. Determination of Catechins and Caffeine
4.9.7. Determination of Morphological Parameters of Tea Leaf
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Axling, U.; Olsson, C.; Xu, J.; Fernandez, C.; Larsson, S.; Strom, K.; Ahrne, S.; Holm, C.; Molin, G.; Berger, K. Green tea powder and Lactobacillus plantarum affect gut microbiota, lipid metabolism and inflammation in high-fat fed C57BL/6J mice. Nutr. Metab. 2012, 9, 105. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, J.; Sheng, J.; Fang, Y.; Li, F.; Sun, G.; Hu, Q. Lnhibition of cyclophoshamide-induced mutagenicity by microsized powder of selenium-enriched green tea in mice. J. Agric. Food Chem. 2008, 56, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Gao, Z.; Li, T.; Song, W.; Xiao, W.; Zheng, J.; Chen, H.; Chen, G.; Zou, H. Anti-tumor activity and the mechanism of a green tea (Camellia sinensis) polysaccharide on prostate cancer. Int. J. Biol. Macromol. 2019, 122, 95–103. [Google Scholar] [CrossRef]
- Yuda, N.; Tanaka, M.; Suzuki, M.; Asano, Y.; Ochi, H.; Iwatsuki, K. Polyphenols extracted from black tea (Camellia Sinensis) residue by hot-compressed water and their inhibitory effect on pancreatic lipase in vitro. J. Food Sci. 2012, 77, H254–H261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, X.; Li, L.; Xu, Z.; Huang, H.; Zhao, J.; Zhang, D.; Yin, X.; Sheng, J.; Tang, J. Pu-erh tea powder preventive effects on cisplatin-induced liver oxidative damage in wistar rats. Asian Pac. J. Cancer Prev. 2014, 15, 7389–7394. [Google Scholar] [CrossRef]
- Zhao, J.; Li, P.; Xia, T.; Wan, X. Exploring plant metabolic genomics: Chemical diversity, metabolic complexity in the biosynthesis and transport of specialized metabolites with the tea plant as a model. Crit. Rev. Biotechnol. 2020, 40, 667–688. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, M.; Li, Y.; Wan, X.; Yang, X. Characterization of the orchid-like aroma contributors in selected premium tea leaves. Food Res. Int. 2020, 129, 108841. [Google Scholar] [CrossRef]
- Yang, Y.; Hua, J.; Deng, Y.; Jiang, Y.; Qian, M.C.; Wang, J.; Li, J.; Zhang, M.; Dong, C.; Yuan, H. Aroma dynamic characteristics during the process of variable-temperature final firing of Congou black tea by electronic nose and comprehensive two- dimensional gas chromatography coupled to time-of-flight mass spectrometry. Food Res. Int. 2020, 137, 109656. [Google Scholar] [CrossRef]
- Flaig, M.; Qi, S.C.; Wei, G.; Yang, X.; Schieberle, P. Characterisation of the key aroma compounds in a Longjing green tea infusion (Camellia sinensis) by the sensomics approach and their quantitative changes during processing of the tea leaves. Eur. Food Res. Technol. 2020, 246, 2411–2425. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef]
- Nakano, S.; Megro, S.-I.; Hase, T.; Suzuki, T.; Isemura, M.; Nakamura, Y.; Ito, S. Computational molecular docking and X-ray crystallographic studies of catechins in new drug design strategies. Molecules 2018, 23, 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Cheng, H.; Xu, P.; Wang, Y. Regulation of biosynthesis of the main flavor-contributing metabolites in tea plant (Camellia sinensis): A review. Crit. Rev. Food Sci. Nutr. 2022, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Ning, W.; Wei, D.; Jiang, M.; Zhu, K.; Wang, X.; Edwards, D.; Odeny, D.A.; Cheng, S. Plant genome resequencing and population genomics: Current status and future prospects. Mol. Plant. 2023, 16, 1252–1268. [Google Scholar] [CrossRef] [PubMed]
- Morrell, P.L.; Buckler, E.S.; Ross-Ibarra, J. Crop genomics: Advances and applications. Nat. Rev. Genet. 2012, 13, 85–96. [Google Scholar] [CrossRef]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant. 2019, 12, 30–43. [Google Scholar] [CrossRef]
- Wu, J.; Wang, L.; Fu, J.; Chen, J.; Wei, S.; Zhang, S.; Zhang, J.; Tang, Y.; Chen, M.; Zhu, J.; et al. Resequencing of 683 common bean genotypes identifies yield component trait associations across a north-south cline. Nat. Genet. 2020, 52, 118–125. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Zuo, H.; Zheng, W.; Zhang, S.; Huang, Y.; Pingcuo, G.; Ying, H.; Zhao, F.; Li, Y.; et al. Genomic basis of high-altitude adaptation in Tibetan Prunus fruit trees. Curr. Biol. 2021, 31, 3848–3860.e8. [Google Scholar] [CrossRef]
- Lovell, J.T.; MacQueen, A.H.; Mamidi, S.; Bonnette, J.; Jenkins, J.; Napier, J.D.; Sreedasyam, A.; Healey, A.; Session, A.; Shu, S.; et al. Genomic mechanisms of climate adaptation in polyploid bioenergy switchgrass. Nature 2021, 590, 438–444. [Google Scholar] [CrossRef]
- Sedivy, E.J.; Wu, F.; Hanzawa, Y. Soybean domestication: The origin, genetic architecture and molecular bases. New Phytol. 2017, 214, 539–553. [Google Scholar] [CrossRef]
- Xia, E.; Tong, W.; Wu, Q.; Wei, S.; Zhao, J.; Zhang, Z.; Wei, C.; Wan, X. Tea plant genomics: Achievements, challenges and perspectives. Hortic. Res. 2020, 7, 7. [Google Scholar] [CrossRef]
- Chen, Y.; Niu, S.; Deng, X.; Song, Q.; He, L.; Bai, D.; He, Y. Genome-wide association study of leaf-related traits in tea plant in Guizhou based on genotyping-by-sequencing. BMC Plant Biol. 2023, 23, 196. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Koiwa, H.; Song, Q.; Qiao, D.; Chen, J.; Zhao, D.; Chen, Z.; Wang, Y.; Zhang, T. Development of core-collections for GuiZhou tea genetic resources and GWAS of leaf size using SNP developed by genotyping-by-sequencing. PeerJ 2020, 8, e8572. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Uchida, T.; Tanaka, Y.; Katai, H.; Nagano, A.J.; Morita, A.; Ikka, T. Genomic predictions and genome-wide association studies based on RAD-seq of quality-related metabolites for the genomics-assisted breeding of tea plants. Sci. Rep. 2020, 10, 17480. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Y.; Yang, P.; Cheng, Y.; Huang, F.; Li, S.; Yang, Y. Population whole-genome resequencing reveals the phylogenetic relationships and population structure of four Hunan typical tea landraces. Bev. Plant Res. 2023, 3, 9. [Google Scholar] [CrossRef]
- Fang, K.; Xia, Z.; Li, H.; Jiang, X.; Qin, D.; Wang, Q.; Wang, Q.; Pan, C.; Li, B.; Wu, H. Genome-wide association analysis identified molecular markers associated with important tea flavor-related metabolites. Hortic. Res. 2021, 8, 42. [Google Scholar] [CrossRef]
- Lu, L.; Chen, H.; Wang, X.; Zhao, Y.; Yao, X.; Xiong, B.; Deng, Y.; Zhao, D. Genome-level diversification of eight ancient tea populations in the Guizhou and Yunnan regions identifies candidate genes for core agronomic traits. Hortic. Res. 2021, 8, 190. [Google Scholar] [CrossRef]
- Niu, S.; Song, Q.; Koiwa, H.; Qiao, D.; Zhao, D.; Chen, Z.; Liu, X.; Wen, X. Genetic diversity, linkage disequilibrium, and population structure analysis of the tea plant (Camellia sinensis) from an origin center, Guizhou plateau, using genome-wide SNPs developed by genotyping-by-sequencing. BMC Plant Biol. 2019, 19, 328. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Gao, X.F.; Yang, J.; Kong, X.R. Genome-wide association study to identify favorable snp allelic variations and candidate genes that control the timing of spring bud flush of tea (Camellia Sinensis) using slaf-seq. J. Agric. Food Chem. 2019, 67, 10380–10391. [Google Scholar] [CrossRef] [PubMed]
- Kottawa-Arachchi, J.D.; Gunasekare, M.T.K.; Ranatunga, M.A.B. Biochemical diversity of global tea Camellia sinensis (L.) O. Kuntze germplasm and its exploitation: A review. Genet. Resour. Crop Evol. 2019, 66, 259–273. [Google Scholar] [CrossRef]
- Kottawa-Arachchi, J.D.; Gunasekare, M.T.K.; Ranatunga, M.A.B.; Punyasiri, P.A.N.; Jayasinghe, L. Use of biochemical compounds in tea germplasm characterization and its implications in tea breeding in Sri Lanka. J. Natl. Sci. Found. Sri Lanka 2013, 41, 309–318. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-c.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef]
- Duan, N.; Bai, Y.; Sun, H.; Wang, N.; Ma, Y.; Li, M.; Wang, X.; Jiao, C.; Legall, N.; Mao, L.; et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 2017, 8, 249. [Google Scholar] [CrossRef]
- Lam, H.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.; Li, M.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef]
- He, L.; Luo, J.; Niu, S.; Bai, D.; Chen, Y. Population structure analysis to explore genetic diversity and geographical distribution characteristics of wild tea plant in Guizhou Plateau. BMC Plant Biol. 2023, 23, 255. [Google Scholar] [CrossRef] [PubMed]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide association studies in maize: Praise and stargaze. Mol. Plant. 2017, 10, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zheng, Z.; Qi, T.; Kemper, K.E.; Wray, N.R.; Visscher, P.M.; Yang, J. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 2019, 51, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, G.; Zhang, A.; Loladze, A.; Hu, Y.; Wang, H.; Qu, J.; Zhang, X.; Olsen, M.; San Vicente, F.; et al. Genome-wide association study and genomic prediction of Fusarium ear rot resistance in tropical maize germplasm. Crop J. 2021, 9, 325–341. [Google Scholar] [CrossRef]
- Zhong, H.; Liu, S.; Sun, T.; Kong, W.; Deng, X.; Peng, Z.; Li, Y. Multi-locus genome-wide association studies for five yield-related traits in rice. BMC Plant Biol. 2021, 21, 364. [Google Scholar] [CrossRef]
- Bai, S.; Hong, J.; Su, S.; Li, Z.; Wang, W.; Shi, J.; Liang, W.; Zhang, D. Genetic basis underlying tiller angle in rice (Oryza sativa L.) by genome-wide association study. Plant Cell Rep. 2022, 41, 1707–1720. [Google Scholar]
- Qin, J.; Wang, F.; Zhao, Q.; Shi, A.; Zhao, T.; Song, Q.; Ravelombola, W.; An, H.; Yan, L.; Yang, C.; et al. Identification of candidate genes and genomic selection for seed protein in soybean breeding pipeline. Front. Plant Sci. 2022, 13, e882723. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Alexander, L.W. Genome-wide association studies for inflorescence type and remontancy in Hydrangea macrophylla. Hortic. Res. 2020, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhong, H.; Meng, X.; Sun, T.; Li, Y.; Pinson, S.R.M.; Chang, S.K.C.; Peng, Z. Genome-wide association studies of ionomic and agronomic traits in USDA mini core collection of rice and comparative analyses of different mapping methods. BMC Plant Biol. 2020, 20, 441. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Zhang, D.; Li, C.; Buckler, E.S.; Li, Y.; Zhang, Z.; Wang, T. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnol. J. 2016, 14, 1551–1562. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Manes, N.; Brauer, E.K.; Hepworth, S.; Subramaniam, R. MAMP and DAMP signaling contributes resistance to Fusarium graminearum in Arabidopsis. J. Exp. Bot. 2021, 72, 6628–6639. [Google Scholar] [CrossRef]
- Rebaque, D.; del Hierro, I.; Lopez, G.; Bacete, L.; Vilaplana, F.; Dallabernardina, P.; Pfrengle, F.; Jorda, L.; Sanchez-Vallet, A.; Perez, R.; et al. Cell wall-derived mixed-linked beta-1,3/1,4-glucans trigger immune responses and disease resistance in plants. Plant J. 2021, 106, 601–615. [Google Scholar] [CrossRef]
- Ahammed, G.J.; Wu, Y.; Wang, Y.; Guo, T.; Shamsy, R.; Li, X. Epigallocatechin-3-Gallate (EGCG): A unique secondary metabolite with diverse roles in plant-environment interaction. Environ. Exp. Bot. 2023, 209, 105299. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, W.; Li, N.; Hao, X.; Wang, L.; Xiao, B.; Wang, X.; Yang, Y. Metabolic changes of caffeine in tea plant (Camellia Sinensis (L.) O. kuntze) as defense response to colletotrichum fructicola. J. Agric. Food Chem. 2016, 64, 6685–6693. [Google Scholar] [CrossRef]
- Jain, N.; Khurana, P.; Khurana, J.P. AtTLP2, a Tubby-like protein, plays intricate roles in abiotic stress signalling. Plant Cell Rep. 2023, 42, 235–252. [Google Scholar] [CrossRef]
- Wardhan, V.; Pandey, A.; Chakraborty, S.; Chakraborty, N. Chickpea transcription factor CaTLP1 interacts with protein kinases, modulates ROS accumulation and promotes ABA-mediated stomatal closure. Sci. Rep. 2016, 6, 38121. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Saito, S.; Endo, K.; Kono, M.; Kakei, T.; Taketa, H.; Kato, M.; Hamamoto, S.; Grenzi, M.; Costa, A.; et al. Green tea catechins, (-)-catechin gallate, and (-)-gallocatechin gallate are potent inhibitors of aba-induced stomatal closure. Adv. Sci. 2022, 9, 2201403. [Google Scholar] [CrossRef] [PubMed]
- Debieu, M.; Huard-Chauveau, C.; Genissel, A.; Roux, F.; Roby, D. Quantitative disease resistance to the bacterial pathogen Xanthomonas campestris involves an Arabidopsis immune receptor pair and a gene of unknown function. Mol. Plant Pathol. 2016, 17, 510–520. [Google Scholar] [CrossRef]
- Miao, R.; Jiang, D.; Musa, A.; Zhou, Q.; Guo, M.; Wang, Y. Effectiveness of shrub planting and grazing exclusion on degraded sandy grassland restoration in Horqin sandy land in Inner Mongolia. Ecol. Eng. 2015, 74, 164–173. [Google Scholar] [CrossRef]
- Lei, X.; Wang, Y.; Zhou, Y.; Chen, Y.; Chen, H.; Zou, Z.; Zhou, L.; Ma, Y.; Chen, F.; Fang, W. TeaPGDB: Tea plant genome database. Bev. Plant Res. 2021, 1, 5. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Ren, S.; Ahmed, N.; Bertels, K.; Al-Ars, Z. GPU accelerated sequence alignment with traceback for GATK HaplotypeCaller. BMC Genom. 2019, 20 (Suppl. S2), 184. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Revell, L.J.; Chamberlain, S.A. Rphylip: An R interface for PHYLIP. Methods Ecol. Evol. 2014, 5, 976–981. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Montana, G.; Hoggart, C. Statistical software for gene mapping by admixture linkage disequilibrium. Brief. Bioinform. 2007, 8, 393–395. [Google Scholar] [CrossRef]
- Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic-structure of a tropical understory shurb, psychotria officinalis (rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGEDI: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Francis, R.M. POPHELPER: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.; Xu, J.; He, W.; Yang, T. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A-Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Gomes, F.E.; Casanova, M.I.; Mouttham, L.; Bannasch, D.L.; Park, S.; Kim, S.; Young, L.J.; Daley, N.L.; Thomasy, S.M.; Castelhano, M.G.; et al. A genome-wide association study to investigate genetic loci associated with primary glaucoma in American Cocker Spaniels. Am. J. Vet. Res. 2022, 83, 1–8. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Yang, B.; Wu, Y.; Zhu, M.; Fan, S.; Lin, J.; Zhang, K.; Li, S.; Chi, H.; Li, Y.; Chen, H.; et al. Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 9, 904–906. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, Y.; Peng, A.; Cui, J.; Zhao, M.; Pan, Y.; Zhang, M.; Tian, K.; Schwab, W.; Song, C. Single-cell transcriptome atlas reveals developmental trajectories and a novel metabolic pathway of catechin esters in tea leaves. Plant Biotechnol. J. 2022, 20, 2089–2106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Porra, R.J. The chequered history of the development and use of simultaneous equations for the accurate determination of chlorophylls a and b. Photosynth. Res. 2002, 73, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Ren, S.; Zhang, Y.; Duan, Y.; Shen, J.; Zhu, X.; Wang, Y.; Ma, Y.; Zou, Z.; Fang, W. Effects of geographic locations and topographical factors on secondary metabolites distribution in green tea at a regional scale. Food Control 2020, 110, 106979. [Google Scholar] [CrossRef]
- Duan, Y.; Shen, J.; Zhang, X.; Wen, B.; Ma, Y.; Wang, Y.; Fang, W.; Zhu, X. Effects of soybean–tea intercropping on soil-available nutrients and tea quality. Acta Physiol. Plant. 2019, 41, 140. [Google Scholar] [CrossRef]
- Xu, J.; Qu, F.; Shen, B.; Huang, Z.; Li, X.; Weng, H.; Ye, D.; Wu, R. Rapid detection of tea polyphenols in fresh tea leaves based on fusion of visible/short-wave and long-wave near infrared spectroscopy and its device development. Appl. Sci. 2023, 13, 1739. [Google Scholar] [CrossRef]
- Wen, B.; Zhang, X.; Ren, S.; Duan, Y.; Zhang, Y.; Zhu, X.; Wang, Y.; Ma, Y.; Fang, W. Characteristics of soil nutrients, heavy metals and tea quality in different intercropping patterns. Agrofor. Syst. 2019, 94, 963–974. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tarit | Gene Locus | Position | p-Value | Exon | SNP | Amino Acid | Gene Annotation |
|---|---|---|---|---|---|---|---|
| GCG | CSS0026218 | 164,656,928 | 6.02 × 10−11 | 1 | GAG-GGG | Glu-Gly | LysM-containing receptor-like kinase |
| CSS0027172 | 140,146,183 | 1.75 × 10−9 | 1 | ACU-UCU | Thr-Ser | tubby-like protein transcription | |
| GC | CSS0044862 | 144,566,106 | 4.57 × 10−10 | 1 | UAU-UGU | Tyr-Cys | plant protein (DUF247) |
| CAF | CSS0039426 | 141,418,417 | 2.38 × 10−9 | 1 | GCT-GGT | Ala-Gly | CREB-regulated transcription coactivator 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Li, H.; Li, P.; Zhang, H.; Han, Z.; Yang, B.; Duan, Y.; Njeri, N.S.; Yang, D.; Zheng, J.; et al. Genome-Wide Association Studies of Biluochun Tea Plant Populations in Dongting Mountain and Comprehensive Identification of Candidate Genes Associated with Core Agronomic Traits by Four Analysis Models. Plants 2023, 12, 3719. https://doi.org/10.3390/plants12213719
Lei X, Li H, Li P, Zhang H, Han Z, Yang B, Duan Y, Njeri NS, Yang D, Zheng J, et al. Genome-Wide Association Studies of Biluochun Tea Plant Populations in Dongting Mountain and Comprehensive Identification of Candidate Genes Associated with Core Agronomic Traits by Four Analysis Models. Plants. 2023; 12(21):3719. https://doi.org/10.3390/plants12213719
Chicago/Turabian StyleLei, Xiaogang, Haoyu Li, Pingping Li, Huan Zhang, Zhaolan Han, Bin Yang, Yu Duan, Ndombi Salome Njeri, Daqiang Yang, Junhua Zheng, and et al. 2023. "Genome-Wide Association Studies of Biluochun Tea Plant Populations in Dongting Mountain and Comprehensive Identification of Candidate Genes Associated with Core Agronomic Traits by Four Analysis Models" Plants 12, no. 21: 3719. https://doi.org/10.3390/plants12213719
APA StyleLei, X., Li, H., Li, P., Zhang, H., Han, Z., Yang, B., Duan, Y., Njeri, N. S., Yang, D., Zheng, J., Ma, Y., Zhu, X., & Fang, W. (2023). Genome-Wide Association Studies of Biluochun Tea Plant Populations in Dongting Mountain and Comprehensive Identification of Candidate Genes Associated with Core Agronomic Traits by Four Analysis Models. Plants, 12(21), 3719. https://doi.org/10.3390/plants12213719