

Molecular Mechanism Underlying the Sorghum sudanense (Piper) Stapf. Response to Osmotic Stress Determined via Single-Molecule Real-Time Sequencing and Next-Generation Sequencing

Abstract
1. Introduction
2. Results
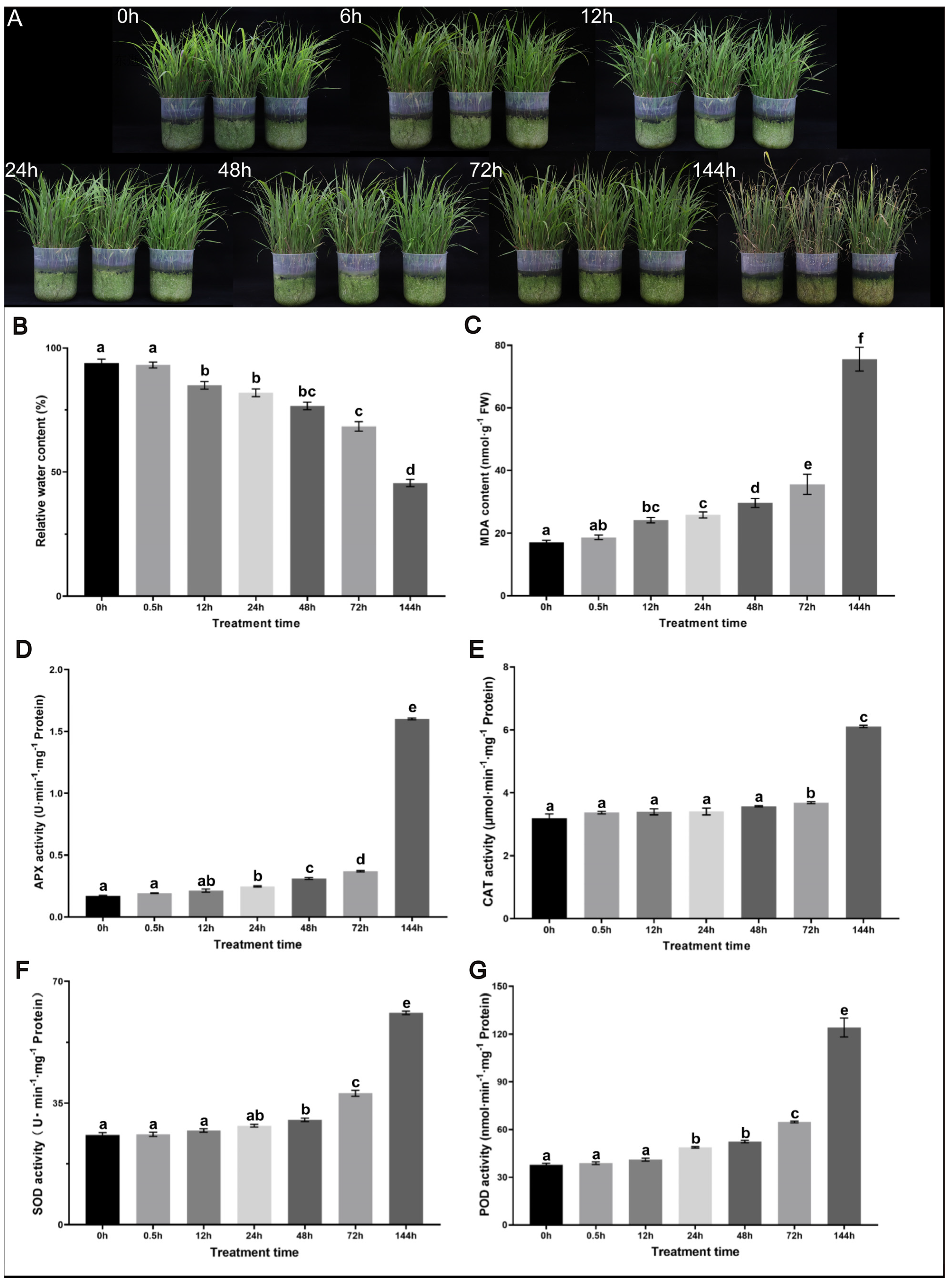
2.1. Phenotypic and Physiological Responses of Sorghum sudanense to Drought Stress
2.2. Full-Length Sequences Obtained via PacBio SMRT Sequencing
2.3. De Novo Assembly of Illumina RNA-Seq Data
2.4. Functional Annotation
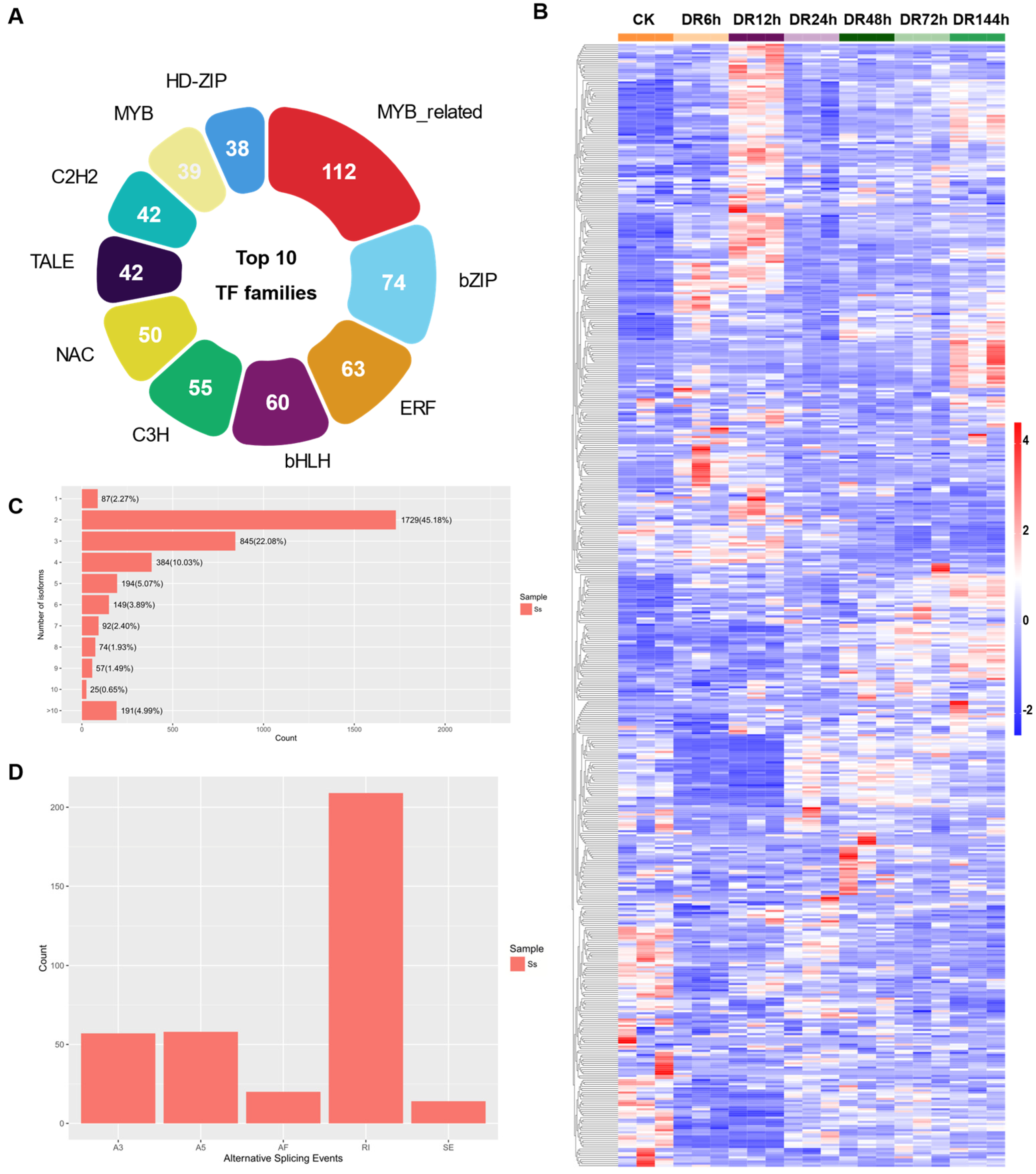
2.5. Transcription Factor Identification and Alternative Splicing Analysis
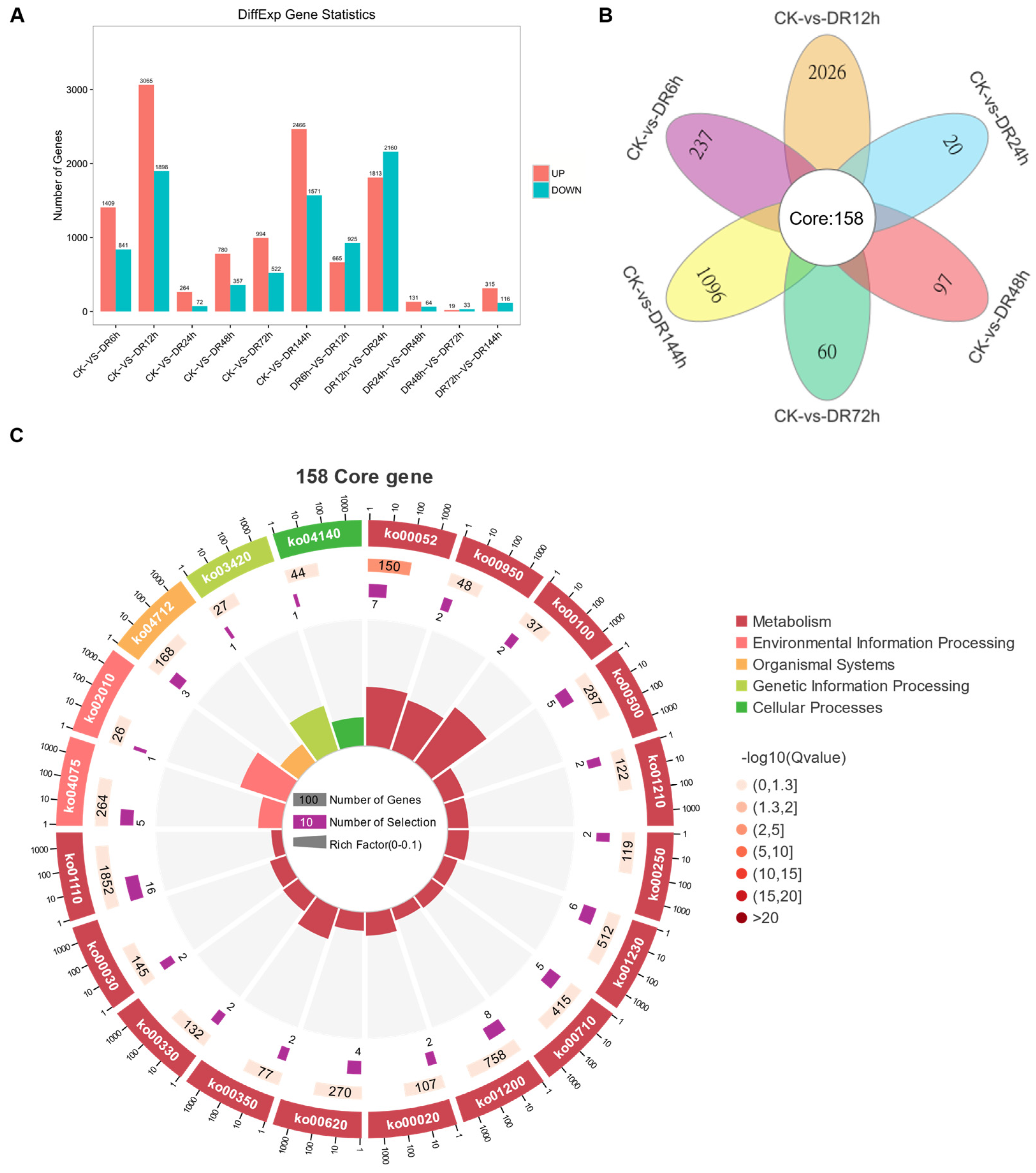
2.6. Identification and Analysis of Differentially Expressed Genes
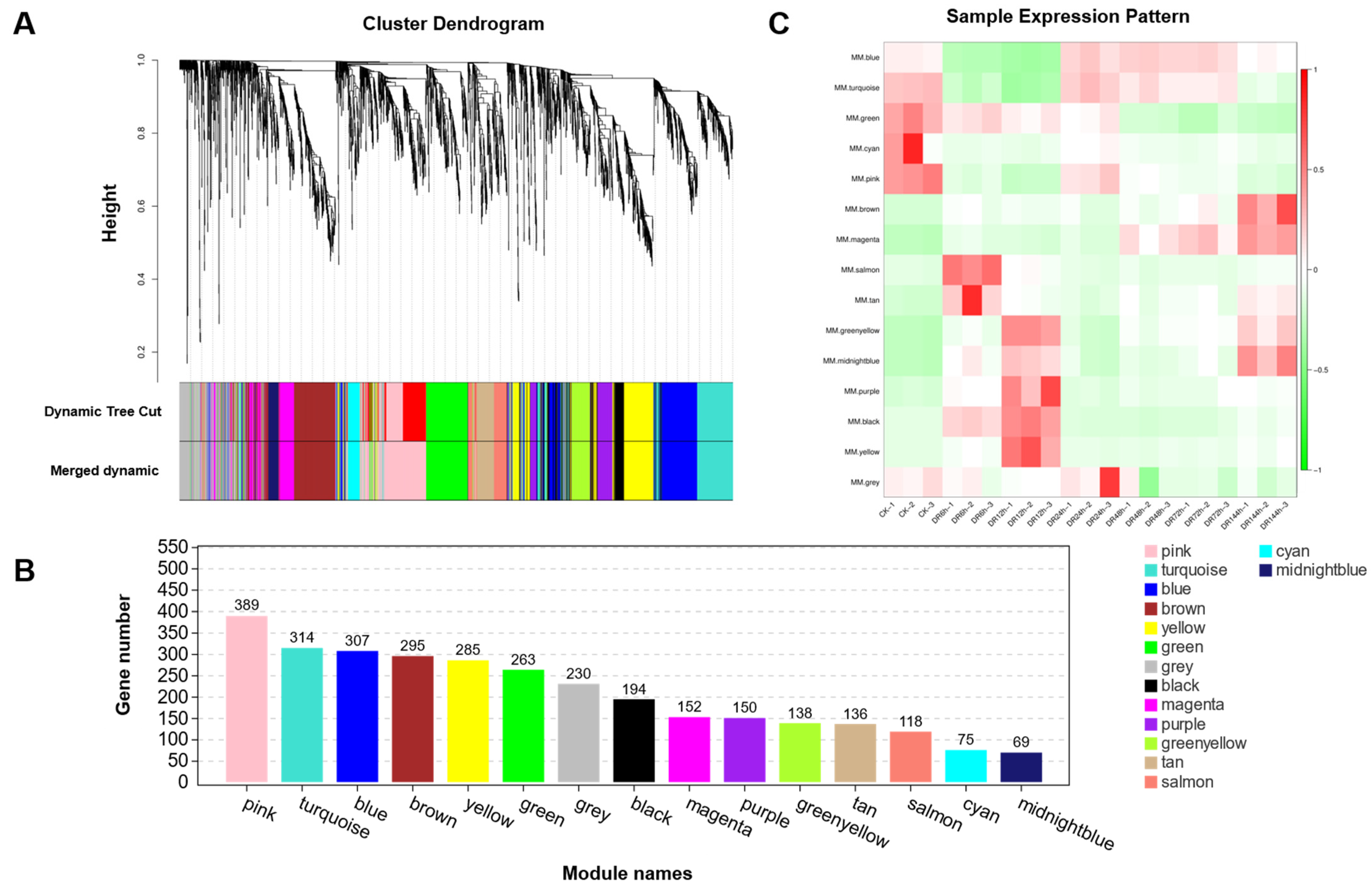
2.7. Weighted Gene Co-Expression Network Analysis
2.8. Validation of Gene Expression Levels
3. Discussion
4. Materials and Methods
4.1. Plant Growth and Collecting
4.2. Physiological Assays
4.3. RNA-Seq Library Construction and Sequencing
4.4. SMRT Library Construction, Sequencing, and Data Processing
4.5. Functionally Annotate Full-Length Transcripts and Identify Differentially Expressed Genes
4.6. Identification of Transcription Factors and Alternative Splicing
4.7. Weighted Gene Co-Expression Network Analysis
4.8. Validation of the Accuracy of Sequencing Results via qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Smith, S.J.; Dillow, D.W.; Young, L.B. Disposition of Fertilizer Nitrate Applied to Sorghum-Sudangrass in the Southern Plains1. J. Environ. Qual. 1982, 11, 341–344. [Google Scholar] [CrossRef]
- Al-Suhaibani, N.A. Effect of Irrigation Intervals and Nitrogen Fertilizer Rates on Fresh Forage Yield of Sudangrass [Sorghum sudanense (Piper) Stapf.]. King Saud Univ. 2006, 142, 5–17. [Google Scholar]
- Zhan, Q.; Qian, Z. Heterosis utilization of hybrid between sorghum [Sorghum bicolor (L.) Moench] and sudangrass [Sorghum sudanense (Piper) Stapf]. Acta Agron. Sin. 2004, 30, 73–77. [Google Scholar]
- Awad, A.; Hafiz, S.; Hammada, M.S.; El-Nouby, A.; El-Hendawy, S. Grain yield production of Sudan grass (Sorghum sudanense (Piper) Stapf) as influenced by cutting numbers, potassium rates, and intrarow spacing in a semiarid environment. Turk. J. Agric. For. 2014, 37, 657–664. [Google Scholar] [CrossRef]
- Bibi, A.; Sadaqat, H.A.; Akram, H.M.; Khan, T.M.; Usman, B.F. Physiological and agronomic responses of Sudangrass to water stress physiological and agronomic responses of sudangrass to water stress. J. Agric. Res. 2010, 48, 369–380. [Google Scholar]
- Li, T.; Zhang, Y.; Liu, Y.; Li, X.; Hao, G.; Han, Q.; Dirk, L.; Downie, A.B.; Ruan, Y.L.; Wang, J. Raffinose synthase enhances drought tolerance through raffinose synthesis or galactinol hydrolysis in maize and Arabidopsis plants. J. Biol. Chem. 2020, 295, 8064–8077. [Google Scholar] [CrossRef]
- Amiard, V.; Morvan-Bertrand, A.; Billard, J.-P.; Huault, C.; Keller, F.; Prud’Homme, M.-P. Fructans, But Not the Sucrosyl-Galactosides, Raffinose and Loliose, Are Affected by Drought Stress in Perennial Ryegrass. Plant Physiol. 2003, 132, 2218–2229. [Google Scholar] [CrossRef]
- Peleg, Z.; Fahima, T.; Krugman, T.; Abbo, S.; Saranga, Y. Genomic dissection of drought resistance in durum wheat x wild emmer wheat recombinant inbreed line population. Plant Cell Environ. 2010, 32, 758–779. [Google Scholar] [CrossRef]
- Egert, A.; Keller, F.; Peters, S. Abiotic stress-induced accumulation of raffinose in Arabidopsis leaves is mediated by a single raffinose synthase (RS5, At5g40390). BMC Plant Biol. 2013, 13, 218. [Google Scholar] [CrossRef]
- Sui, X.L.; Meng, F.Z.; Wang, H.Y.; Wei, Y.X.; Li, R.F.; Wang, Z.Y.; Hu, L.P.; Wang, S.H.; Zhang, Z.X. Molecular cloning, characteristics and low temperature response of raffinose synthase gene in Cucumis sativus L. J. Plant Physiol. 2012, 169, 1883–1891. [Google Scholar] [CrossRef]
- Zhao, H.; Gao, Z.; Wang, L.; Wang, J.; Wang, S.; Fei, B.; Chen, C.; Shi, C.; Liu, X.; Zhang, H.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). GigaScience 2018, 7, giy115. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. Transcriptome: Connecting the genome to gene function. Nat. Educ. 2008, 1, 195. [Google Scholar]
- Chen, X.; Liu, X.; Zhu, S.; Tang, S.; Mei, S.; Chen, J.; Li, S.; Liu, M.; Gu, Y.; Dai, Q.; et al. Transcriptome-referenced association study of clove shape traits in garlic. DNA Res. 2018, 25, 587–596. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Schaarschmidt, S.; Fischer, A.; Lawas, L.; Alam, R.; Septiningsih, E.M.; Bailey-Serres, J.; Jagadish, S.; Huettel, B.; Hincha, D.K.; Zuther, E. Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. Int. J. Mol. Sci. 2020, 21, 8148. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, F.; Shuai, Y.; Huang, L.; Zhang, X. Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum. Int. J. Mol. Sci. 2022, 23, 7921. [Google Scholar] [CrossRef]
- Scott, I.M.; Clarke, S.M.; Wood, J.E.; Mur, L.A. Salicylate accumulation inhibits growth at chilling temperature in Arabidopsis. Plant Physiol. 2004, 135, 1040–1049. [Google Scholar] [CrossRef]
- El-Ezz, S.; Taha, A.A.; El-Hadidi, E. Phytoremediation of Some Polluted Soils by Sudan Grass (Sorghum Sodanese L.). Bull. Fac. Eng. Mansoura Univ. 2015, 40, 1. [Google Scholar]
- Zhu, Y.; Wang, X.; Huang, L.; Lin, C.; Zhang, X.; Xu, W.; Peng, J.; Li, Z.; Yan, H.; Luo, F.; et al. Transcriptomic Identification of Drought-Related Genes and SSR Markers in Sudan Grass Based on RNA-Seq. Front. Plant Sci. 2017, 8, 687. [Google Scholar] [CrossRef]
- Castorina, G.; Domergue, F.; Chiara, M.; Zilio, M.; Persico, M.; Ricciardi, V.; Horner, D.S.; Consonni, G. Drought-Responsive ZmFDL1/MYB94 Regulates Cuticle Biosynthesis and Cuticle-Dependent Leaf Permeability. Plant Physiol. 2020, 184, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Huang, X.; Ding, L.; Wang, Z.; Tang, D.; Chen, B.; Ao, L.; Liu, Y.; Kang, Z.; Mao, H. TaERF87 and TaAKS1 synergistically regulate TaP5CS1/TaP5CR1-mediated proline biosynthesis to enhance drought tolerance in wheat. New Phytol. 2023, 237, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wei, Z.; Zhai, H.; Xing, S.; Wang, Y.; He, S.; Gao, S.; Zhao, N.; Zhang, H.; Liu, Q. The IbPYL8-IbbHLH66-IbbHLH118 complex mediates the abscisic acid-dependent drought response in sweet potato. New Phytol. 2022, 236, 2151–2171. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Seymour, G.B.; Lu, C.; Hu, Z.; Chen, X.; Chen, G. An ethylene response factor (ERF5) promoting adaptation to drought and salt tolerance in tomato. Plant Cell Rep. 2012, 31, 349–360. [Google Scholar] [CrossRef]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts. I. Kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys. 1968, 125, 189–198. [Google Scholar] [CrossRef]
- Wang, J.; Sun, P.P.; Chen, C.L.; Wang, Y.; Fu, X.Z.; Liu, J.H. An arginine decarboxylase gene PtADC from Poncirus trifoliata confers abiotic stress tolerance and promotes primary root growth in Arabidopsis. J. Exp. Bot. 2011, 62, 2899–2914. [Google Scholar] [CrossRef]
- Phimchan, P.; Chanthai, S.; Bosland, P.W.; Techawongstien, S. Enzymatic changes in phenylalanine ammonia-lyase, cinnamic-4-hydroxylase, capsaicin synthase, and peroxidase activities in capsicum under drought stress. J. Agric. Food Chem. 2014, 62, 7057–7062. [Google Scholar] [CrossRef]
- Qian, P.; Sun, R.; Basharat, B.; Bullet, A.; Zhou, W. Effects of Hydrogen Sulfide on Growth, Antioxidative Capacity, and Ultrastructural Changes in Oilseed Rape Seedlings Under Aluminum Toxicity. J. Plant Growth Regul. 2014, 33, 526–538. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Mano, J.; Ohno, C.; Domae, Y.; Asada, K. Chloroplastic ascorbate peroxidase is the primary target of methylviologen-induced photooxidative stress in spinach leaves: Its relevance to monodehydroascorbate radical detected with in vivo ESR. Biochim. Biophys. Acta 2001, 1504, 275–287. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e132628. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Read Number | Base Number | Average Length |
|---|---|---|---|
| raw data | 23,326,110 | 32,260,010,130 | 1383 |
| CCS reads | 327,570 | 570,995,745 | 1743 |
| high-quality isoforms | 24,283 | - | - |
| low-quality isoforms | 46 | - | - |
| corrected using NGS data | 24,306 | 39,769,457 | 1636 |
| isoform | 20,199 | 32,881,692 | 1628 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Wang, F.; Xu, Y.; Lin, C.; Li, X.; Xu, W.; Wang, H.; Zhu, Y. Molecular Mechanism Underlying the Sorghum sudanense (Piper) Stapf. Response to Osmotic Stress Determined via Single-Molecule Real-Time Sequencing and Next-Generation Sequencing. Plants 2023, 12, 2624. https://doi.org/10.3390/plants12142624
Liu Q, Wang F, Xu Y, Lin C, Li X, Xu W, Wang H, Zhu Y. Molecular Mechanism Underlying the Sorghum sudanense (Piper) Stapf. Response to Osmotic Stress Determined via Single-Molecule Real-Time Sequencing and Next-Generation Sequencing. Plants. 2023; 12(14):2624. https://doi.org/10.3390/plants12142624
Chicago/Turabian StyleLiu, Qiuxu, Fangyan Wang, Yalin Xu, Chaowen Lin, Xiangyan Li, Wenzhi Xu, Hong Wang, and Yongqun Zhu. 2023. "Molecular Mechanism Underlying the Sorghum sudanense (Piper) Stapf. Response to Osmotic Stress Determined via Single-Molecule Real-Time Sequencing and Next-Generation Sequencing" Plants 12, no. 14: 2624. https://doi.org/10.3390/plants12142624
APA StyleLiu, Q., Wang, F., Xu, Y., Lin, C., Li, X., Xu, W., Wang, H., & Zhu, Y. (2023). Molecular Mechanism Underlying the Sorghum sudanense (Piper) Stapf. Response to Osmotic Stress Determined via Single-Molecule Real-Time Sequencing and Next-Generation Sequencing. Plants, 12(14), 2624. https://doi.org/10.3390/plants12142624