

Exploration of the Regulatory Pathways and Key Genes Involved in the Response to Saline–Alkali Stress in Betula platyphylla via RNA-Seq Analysis


Abstract
1. Introduction
2. Results
2.1. Quality Control of the RNA-Seq Data
2.2. Identification of DEGs
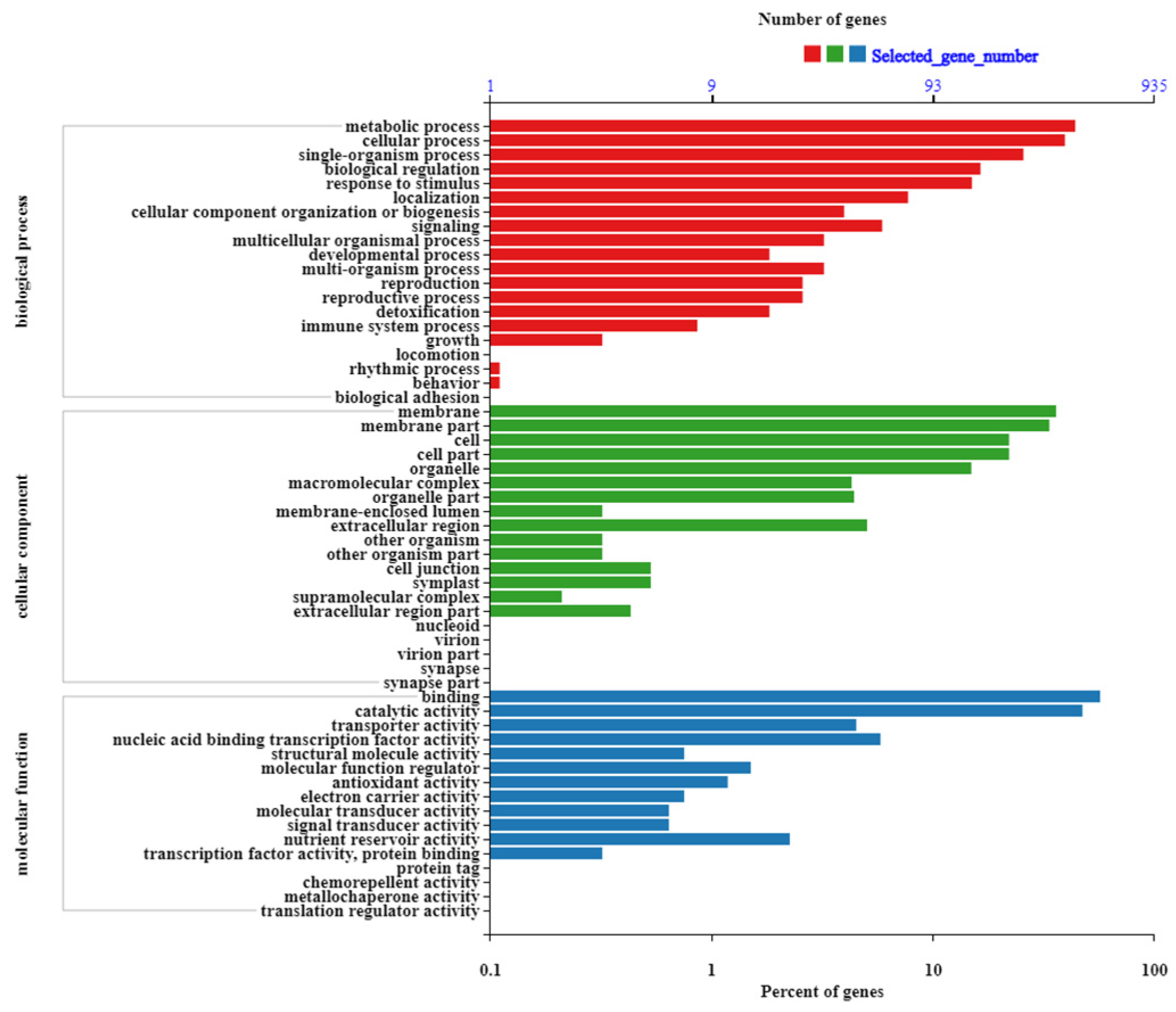
2.3. GO Enrichment Analysis and Functional Annotation of DEGs
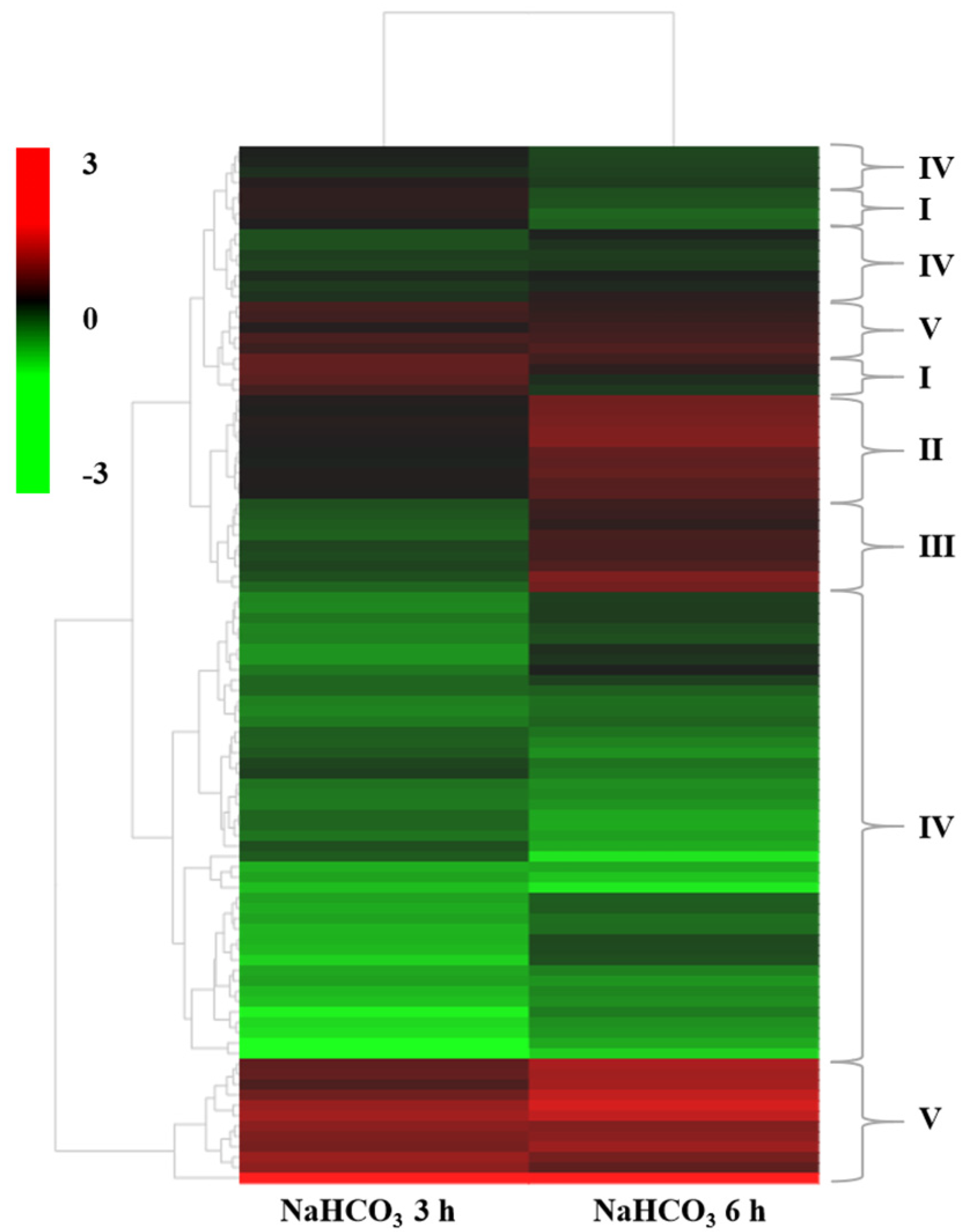
2.4. Hierarchical Clustering Analysis of DEGs
2.5. SNP Analysis of RNA-Seq Data
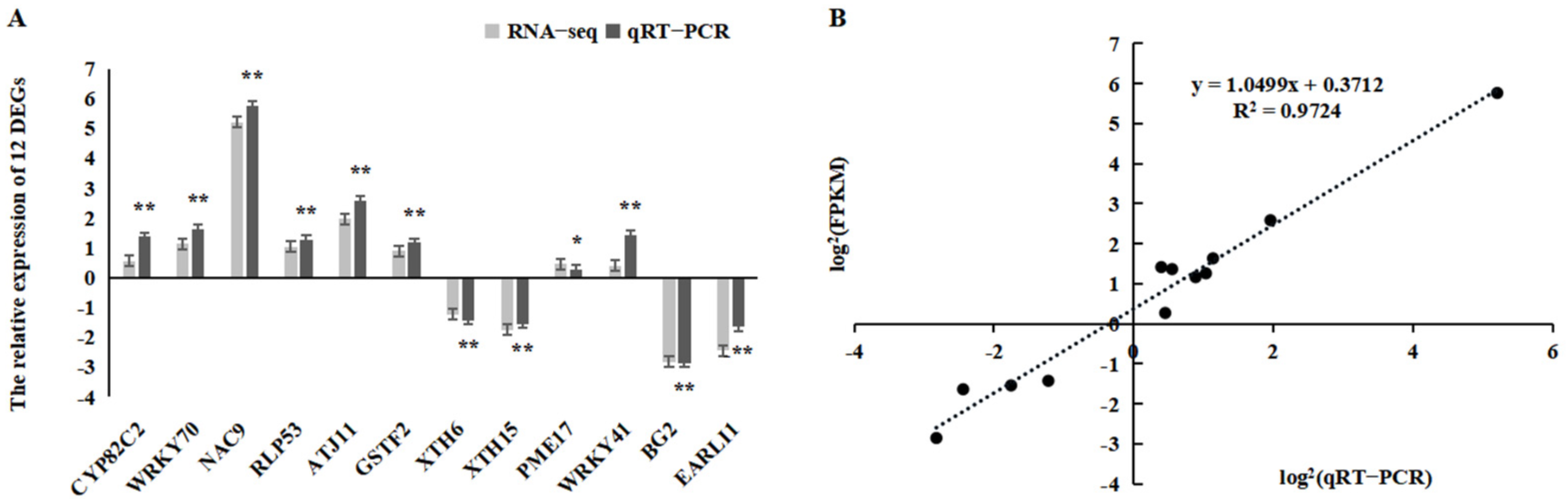
2.6. Confirmation of RNA-Seq Data Using qRT-PCR Analysis
2.7. Oxidative Stress and Cell Membrane Damage Were Alleviated in Plants Overexpressing WRKY70 and NAC9
2.8. Physiological Characterization of WRKY70- and NAC9-Overexpressing Plants
2.9. Expression Changes of Target Genes in WRKY70- and NAC9-Overexpressing Plants
3. Discussion
4. Materials and Methods
4.1. Plant Growth and Stress Treatments
4.2. RNA Extraction, Library Construction, and RNA-Seq
4.3. RNA-Seq Analysis Using Reference Genome-Based Read Mapping
4.4. Analysis of DEGs
4.5. Gene Functional Annotation
4.6. SNP Analysis
4.7. Quantitative Real-Time PCR (qRT-PCR) Verification
4.8. Plasmid Construct Containing TF Genes and Plant Transformation
4.9. Relative Expression and Stress Tolerance Analysis of WRKY70 and NAC9-Overexpressing Plants
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yan, S.; Chong, P.; Zhao, M.; Liu, H. Physiological response and proteomics analysis of Reaumuria soongorica under salt stress. Sci. Rep. 2022, 12, 2539. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Zhao, S.; Fang, Q.; Ruan, J.; Li, S.; Liu, T.; Qi, Y.; Zhang, L.; Zhang, X.; et al. Overexpression of the aldehyde dehydrogenase AhALDH3H1 from Arachis hypogaea in soybean increases saline-alkali stress tolerance. Front. Plant Sci. 2023, 14, 1165384. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Hou, X.; Liang, X. Response Mechanisms of Plants Under Saline-Alkali Stress. Front. Plant Sci. 2021, 12, 667458. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Wang, D. Effects of various salt-alkaline mixed stresses on Aneurolepidium chinense (Trin.) Kitag. Plant Soil 2005, 271, 15–26. [Google Scholar] [CrossRef]
- Gazala, M.A.; Charlotte, E.S.; Al-Azzawi, M.; Timothy, J.F. The effect of combined salinity and waterlogging on the halophyte Suaeda maritima: The role of antioxidants. Environ. Exp. Bot. 2013, 87, 120–125. [Google Scholar]
- Rao, Y.; Peng, T.; Xue, S. Mechanisms of plant saline-alkaline tolerance. J. Plant Physiol. 2023, 281, 153916. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zheng, W.; Tian, Y.; Wu, Y.; Zhou, D.W. Effects of various mixed salt-alkaline stresses on growth, photosynthesis, and photosynthetic pigment concentrations of medicago ruthenica seedlings. Photosynthetica 2011, 49, 275–284. [Google Scholar] [CrossRef]
- Wang, C.; Gao, C.; Wang, L.; Zheng, L.; Yang, C.; Wang, Y. Comprehensive transcriptional profiling of NaHCO3-stressed Tamarix hispida roots reveals networks of responsive genes. Plant Mol. Biol. 2013, 84, 145–157. [Google Scholar] [CrossRef]
- Jiang, Y.; Deyholos, M.K. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006, 12, 6–25. [Google Scholar]
- Wang, P.Y.; Wang, J.X.; Sun, X.M.; Yang, X.; Sun, S.L.; Han, X.; Li, D.D.; Wang, Y.C. Construction of a Hierarchical Gene Regulatory Network to Reveal the Drought Tolerance Mechanism of Shanxin Poplar. Int. J. Mol. Sci. 2022, 24, 384. [Google Scholar] [CrossRef]
- Wicker, T.; Schlagenhauf, E.; Graner, A.; Close, T.J.; Keller, B.; Stein, N. 454 sequencing put to the test using the complex genome of barley. BMC Genom. 2006, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Kitao, M.; Lei, T.T.; Nakamura, T.; Koike, T. Manganese toxicity as indicated by visible foliar symptoms of Japanese white birch (Betula platyphylla var. japonica). Environ. Pollut. 2001, 111, 89–94. [Google Scholar] [CrossRef]
- Pamidi, T.R.K.; Johansson, Ö.; Löfqvist, T.; Shankar, V. Comparison of two different ultrasound reactors for the treatment of cellulose fibers. Ultrason. Sonochem. 2020, 62, 104841. [Google Scholar] [CrossRef]
- Xing, L.; Liu, X.M. Characterization of Betula platyphylla gene transcripts associated with early development of male inflorescence. Mol. Biol. Rep. 2012, 39, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Cui, X.; Lin, S.; Gan, S.; Xing, H.; Dou, D. GmCYP82A3, a Soybean Cytochrome P450 Family Gene Involved in the Jasmonic Acid and Ethylene Signaling Pathway, Enhances Plant Resistance to Biotic and Abiotic Stresses. PLoS ONE 2016, 11, e0162253. [Google Scholar] [CrossRef]
- Zhao, J.L.; Zou, L.; Zhong, L.Y.; Peng, L.X.; Ying, P.L.; Tan, M.L. Effects of polysaccharide elicitors from endophytic Bionectriapityrodes Fat6 on the growth and flavonoid production in tartary buckwheat sprout cultures. Cereal Res. Commun. 2015, 43, 661–671. [Google Scholar] [CrossRef]
- Yang, Y.; Al-Baidhani, H.H.J.; Harris, J.; Riboni, M.; Li, Y.; Mazonka, I.; Bazanova, N.; Chirkova, L.; Sarfraz Hussain, S.; Hrmova, M.; et al. DREB/CBF expression in wheat and barley using the stress-inducible promoters of HD-Zip I genes: Impact on plant development, stress tolerance and yield. Plant Biotechnol. J. 2020, 18, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Geng, Z.; Zhang, C.; Wang, K.; Jiang, X. Whole-genome characterization of Rosa chinensis AP2/ERF transcription factors and analysis of negative regulator RcDREB2B in Arabidopsis. BMC Genom. 2021, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Luo, W.; Ge, M.; Guan, Y.; Tang, Y.; Chen, W.; Lv, J. A Group I WRKY Gene, TaWRKY133, Negatively Regulates Drought Resistance in Transgenic Plants. Int. J. Mol. Sci. 2022, 23, 12026. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Son, S.; Lee, K.S.; Park, Y.J.; Suh, E.J.; Lee, S.I.; Park, S.R. OsWRKY114 Negatively Regulates Drought Tolerance by Restricting Stomatal Closure in Rice. Plants 2022, 11, 1938. [Google Scholar] [CrossRef]
- Liu, W.; Liang, X.; Cai, W.; Wang, H.; Liu, X.; Cheng, L.; Song, P.; Luo, G.; Han, D. Isolation and Functional Analysis of VvWRKY28, a Vitis vinifera WRKY Transcription Factor Gene, with Functions in Tolerance to Cold and Salt Stress in Transgenic Arabidopsis thaliana. Int. J. Mol. Sci. 2022, 23, 13418. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Liu, S.; Zhang, C.; He, J.; Ma, D.; Wang, X.; Dong, T.; Guo, F.; Cai, J.; Long, T.; et al. The unique sweet potato NAC transcription factor IbNAC3 modulates combined salt and drought stresses. Plant Physiol. 2023, 191, 747–771. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jiang, T.; Zhang, Y.; Zhang, K.; Feng, K.; Wu, P.; Li, L. Identification of the NAC Transcription Factors and Their Function in ABA and Salinity Response in Nelumbo nucifera. Int. J. Mol. Sci. 2022, 23, 12394. [Google Scholar] [CrossRef]
- Xi, Y.; Ling, Q.; Zhou, Y.; Liu, X.; Qian, Y. ZmNAC074, a maize stress-responsive NAC transcription factor, confers heat stress tolerance in transgenic Arabidopsis. Front. Plant Sci. 2022, 13, 986628. [Google Scholar] [CrossRef]
- Liu, J.X.; Wu, B.; Feng, K.; Li, M.Y.; Duan, A.Q.; Shen, D.; Yin, L.; Xu, Z.S.; Xiong, A.S. A celery transcriptional repressor AgERF8 negatively modulates abscisic acid and salt tolerance. Mol. Genet. Genom. 2021, 296, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guo, J.; Chen, X.; Zhou, Y.; Pei, Y.; Chen, L.; Ul Haq, S.; Lu, M.; Gong, H.; Chen, R. Pepper bHLH transcription factor CabHLH035 contributes to salt tolerance by modulating ion homeostasis and proline biosynthesis. Hortic. Res. 2022, 9, uhac203. [Google Scholar] [CrossRef]
- Wang, F.Z.; Wang, Q.B.; Kwon, S.Y.; Kwak, S.S.; Su, W.A. Enhanced drought tolerance of transgenic rice plants expressing a pea manganese superoxide dismutase. J. Plant. Physiol. 2005, 162, 465–472. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Meng, H.; Wen, H.; Fan, Y.; Zhao, J. Maize ABP9 enhances tolerance to multiple stresses in transgenic Arabidopsis by modulating ABA signaling and cellular levels of reactive oxygen species. Plant Mol. Biol. 2011, 75, 365–378. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sun, S.L.; Wang, B.; Jiang, Q.; Li, Z.R.; Jia, S.T.; Wang, Y.C.; Guo, H.Y. Genome-wide analysis of BpDof genes and the tolerance to drought stress in birch (Betula platyphylla). PeerJ 2021, 9, e11938. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ahn, J.W.; Jin, U.H.; Choi, D.; Paek, K.H.; Pai, H.S. Activation of the programmed cell death pathway by inhibition of proteasome function in plants. J. Biol. Chem. 2003, 278, 19406–19415. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, X.; Nie, X.; Qu, M.; Zheng, L.; Tan, Z.; Zhao, H.; Huo, L.; Liu, S.; Zhang, B.; et al. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New Phytol. 2015, 207, 692–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, L.; Zhang, C.; Wang, Y.; Lu, M.; Gao, C. ThWRKY4 from Tamarix hispida Can Form Homodimers and Heterodimers and Is Involved in Abiotic Stress Responses. Int. J. Mol. Sci. 2015, 16, 27097–27106. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Number of Genes | |
|---|---|---|
| Control versus 3 h | Control versus 6 h | |
| Total | 595 | 607 |
| Up-regulated | 290 | 199 |
| Down-regulated | 305 | 408 |
| Names | SNP Number | Genic SNP | Intergenic SNP | Transition | Transversion | Heterozygosity |
|---|---|---|---|---|---|---|
| Control | 277,460 | 242,456 | 35,005 | 61.32% | 38.68% | 61.71% |
| NaHCO3 3 h | 275,847 | 241,463 | 34,383 | 61.30% | 38.70% | 61.55% |
| NaHCO3 6 h | 262,684 | 231,583 | 31,101 | 61.30% | 38.70% | 61.80% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, J.; Sun, H.; Zhou, X.; Guo, H.; Wang, Y. Exploration of the Regulatory Pathways and Key Genes Involved in the Response to Saline–Alkali Stress in Betula platyphylla via RNA-Seq Analysis. Plants 2023, 12, 2435. https://doi.org/10.3390/plants12132435
Xue J, Sun H, Zhou X, Guo H, Wang Y. Exploration of the Regulatory Pathways and Key Genes Involved in the Response to Saline–Alkali Stress in Betula platyphylla via RNA-Seq Analysis. Plants. 2023; 12(13):2435. https://doi.org/10.3390/plants12132435
Chicago/Turabian StyleXue, Jukun, Hu Sun, Xuemei Zhou, Huiyan Guo, and Yucheng Wang. 2023. "Exploration of the Regulatory Pathways and Key Genes Involved in the Response to Saline–Alkali Stress in Betula platyphylla via RNA-Seq Analysis" Plants 12, no. 13: 2435. https://doi.org/10.3390/plants12132435
APA StyleXue, J., Sun, H., Zhou, X., Guo, H., & Wang, Y. (2023). Exploration of the Regulatory Pathways and Key Genes Involved in the Response to Saline–Alkali Stress in Betula platyphylla via RNA-Seq Analysis. Plants, 12(13), 2435. https://doi.org/10.3390/plants12132435