

Transcriptional Interactions of Single B-Subgenome Chromosome with C-Subgenome in B. oleracea-nigra Additional Lines


Abstract
1. Introduction
2. Results
2.1. DEGs Groups: MAALs vs. CC; Non-MAALs vs. CC
2.2. Cis- and Trans-Effects on Gene Expression: Dysregulated Genes in Single B-Subgenome Chromosome and C-Subgenome
2.3. Trans-Effect Dysregulation Genes Associated with Alien Chromosomes, and Their Different Transcriptional Responses to Aneuploidy
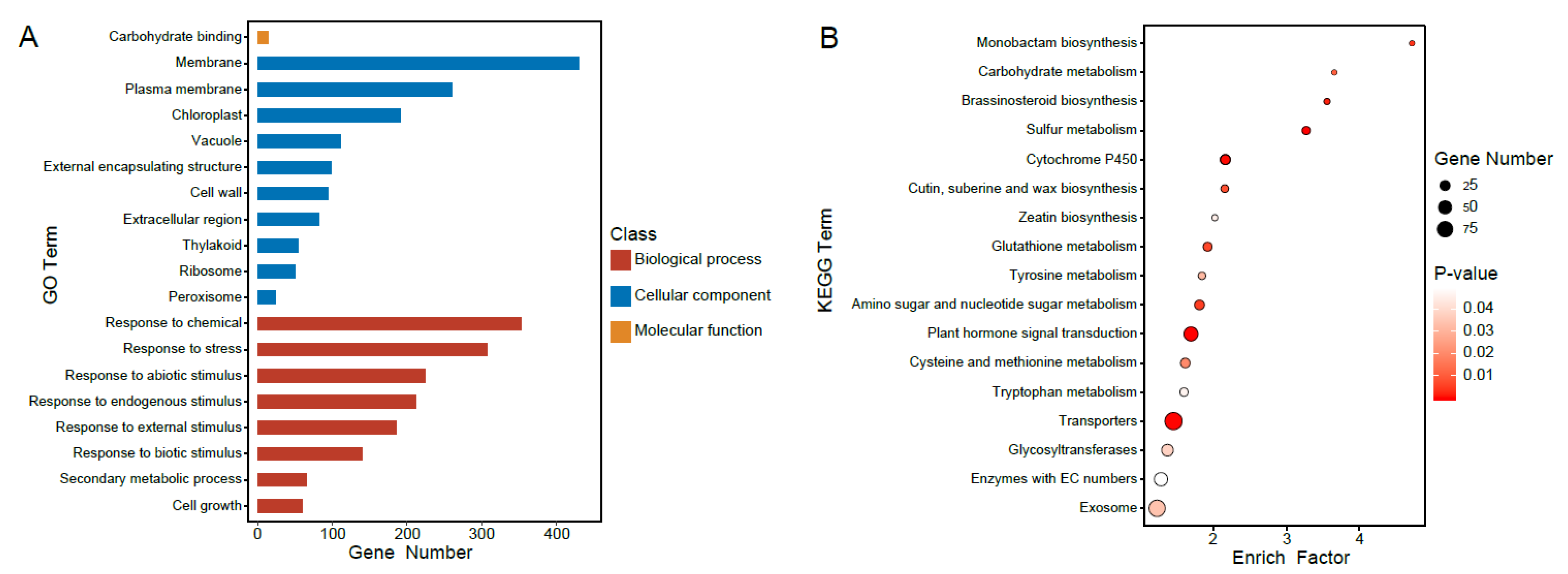
2.4. Functional Analysis of Differently Regulated Gene Groups
2.5. Expression Analysis of Orthologous Genes from MAALs in Parental Genomes
2.6. Bias to Downregulation for Different Levels of Expressed Genes and Their Maintenance
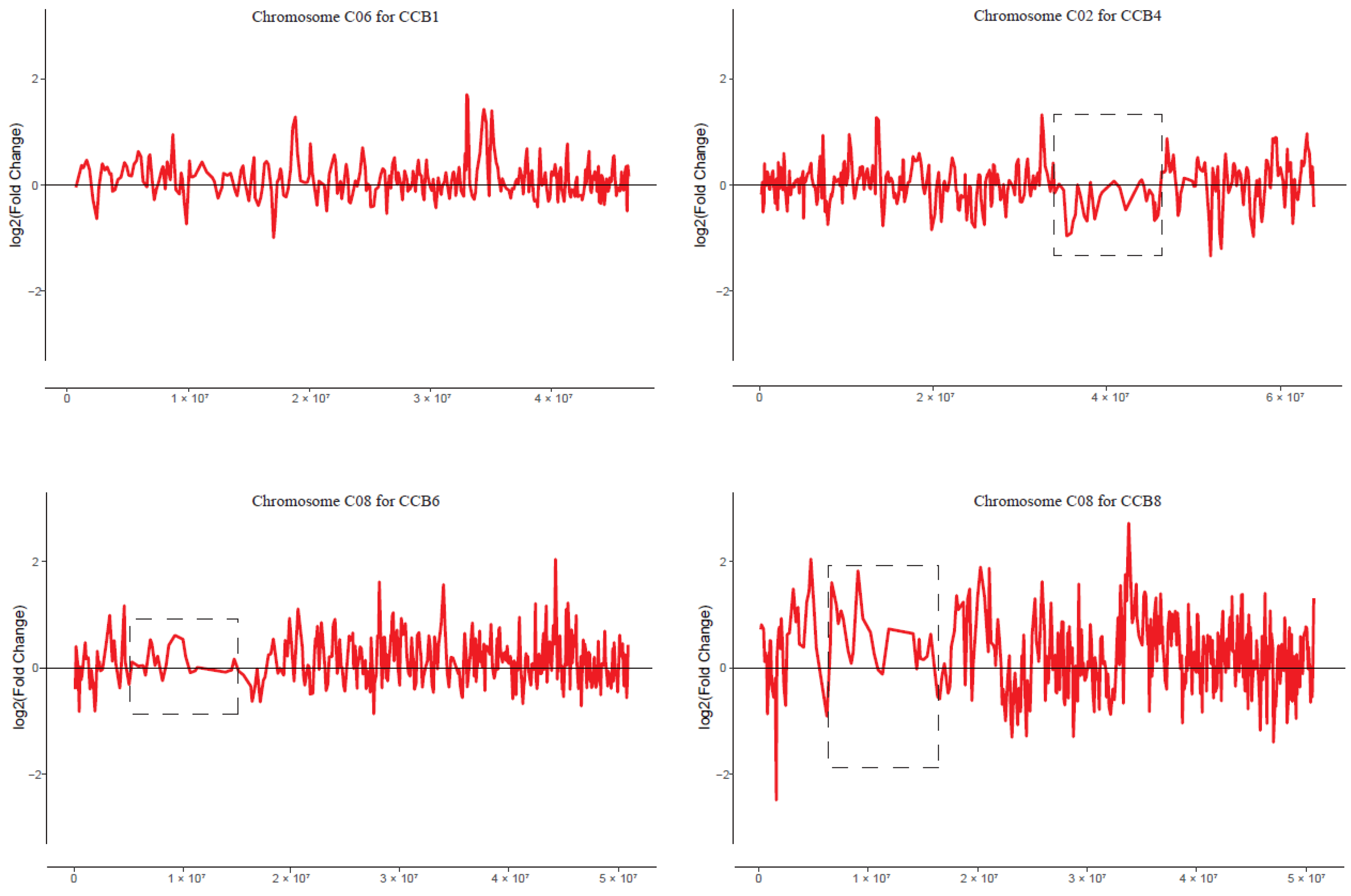
2.7. No Obvious Dysregulation Domains in MAALs
2.8. Validation of DEGs by qRT-PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and RNA-Seq
4.3. Transcriptome Analysis
4.4. Quantitative RT–PCR
4.5. The GO and KEGG Enrichment Analysis
4.6. Data Statistics and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaeta, R.T.; Chris Pires, J. Homoeologous recombination in allopolyploids: The polyploid ratchet. New Phytol. 2009, 186, 18–28. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Last, R.L.; Pichersky, E. Harnessing plant trichome biochemistry for the production of useful compounds. Plant J. 2008, 54, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef]
- Siegel, J.J.; Amon, A. New insights into the troubles of aneuploidy. Annu. Rev. Cell Dev. Biol. 2012, 28, 189–214. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Kerber, E.R. Wheat: Reconstitution of the tetraploid component (AABB) of hexaploids. Science 1964, 143, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Pelé, A.; Trotoux, G.; Eber, F.; Lodé, M.; Gilet, M.; Deniot, G.; Falentin, C.; Nègre, S.; Morice, J.; Rousseau-Gueutin, M.; et al. The poor lonesome A subgenome of Brassica napus var. Darmor (AACC) may not survive without its mate. New Phytol. 2016, 213, 1886–1897. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, B.; Qi, B.; Gou, X.; Dong, Y.; Xu, C.; Zhang, B.; Huang, W.; Liu, C.; Wang, X.; et al. Evolution of the BBAA component of bread wheat during its history at the allohexaploid level. Plant Cell 2014, 26, 2761–2776. [Google Scholar] [CrossRef]
- Zhu, B.; Tu, Y.; Zeng, P.; Ge, X.; Li, Z. Extraction of the constituent subgenomes of the natural allopolyploid rapeseed (Brassica napus L.). Genetics 2016, 204, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Sears, E.R. The aneuploids of common wheat. Mo. Agric. Exp. Stat. Res. Bull. 1954, 572, 1–59. [Google Scholar]
- Zhu, B.; Pan, Q.; Huo, D.; Zeng, P.; Cai, B.; Ge, X.; Li, Z. Transcriptional aneuploidy responses of Brassica rapa-oleracea monosomic alien addition lines (MAALs) derived from natural allopolyploid B. napus. Front. Genet. 2019, 10, 67. [Google Scholar]
- Birchler, J.A.; Bhadra, U.; Bhadra, M.P.; Auger, D.L. Dosage-dependent gene regulation in multicellular eukaryotes: Implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev. Biol. 2001, 234, 275–288. [Google Scholar] [CrossRef]
- Birchler, J.A.; Newton, K.J. Modulation of protein levels in chromosomal dosage series of maize: The biochemical basis of aneuploid syndromes. Genetics 1981, 99, 247–266. [Google Scholar] [CrossRef]
- Birchler, J.A.; Veitia, R.A. The gene balance hypothesis: From classical genetics to modern genomics. Plant Cell 2007, 19, 395–402. [Google Scholar] [CrossRef]
- Huettel, B.; Kreil, D.P.; Matzke, M.; Matzke, A.J.M. Effects of aneuploidy on genome structure, expression, and interphase organization in Arabidopsis thaliana. PLoS Genet. 2008, 4, e1000226. [Google Scholar] [CrossRef]
- Makarevitch, I.; Harris, C. Aneuploidy causes tissue-specific qualitative changes in global gene expression patterns in maize. Plant Physiol. 2010, 152, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, A.; Santoni, F.A.; Bonilla, X.; Sailani, M.R.; Gonzalez, D.; Kind, J.; Chevalier, C.; Thurman, R.; Sandstrom, R.S.; Hibaoui, Y.; et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 2014, 508, 345–350. [Google Scholar] [CrossRef]
- Tan, C.; Pan, Q.; Cui, C.; Xiang, Y.; Ge, X.; Li, Z. Genome-wide gene/genome dosage imbalance regulates gene expressions in synthetic Brassica napus and derivatives (AC, AAC, CCA, CCAA). Front. Plant Sci. 2016, 7, 1432. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Wang, T.; Fu, W.; Harun, A.; Ge, X.; Li, Z. Dosage-dependent gynoecium development and gene expression in Brassica napus-Orychophragmus violaceus addition lines. Plants 2021, 10, 1766. [Google Scholar] [CrossRef]
- Shi, X.; Ng, D.W.; Zhang, C.; Comai, L.; Ye, W.; Chen, Z.J. Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3, 950. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2012, 13, 59–69. [Google Scholar] [CrossRef]
- Guo, M.; Birchler, J.A. Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 1994, 266, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.H.; Cho, D.-Y.; Mattiuzzo, N.R.; Artieri, C.G.; Jiang, L.; Dale, R.K.; Smith, H.E.; McDaniel, J.; Munro, S.; Salit, M.; et al. Mediation of Drosophila autosomal dosage effects and compensation by network interactions. Genome Biol. 2012, 13, R28. [Google Scholar] [CrossRef]
- Zhang, A.; Li, N.; Gong, L.; Gou, X.; Wang, B.; Deng, X.; Li, C.; Dong, Q.; Zhang, H.; Liu, B. Global analysis of gene expression in response to whole-chromosome aneuploidy in hexaploid Wheat. Plant Physiol. 2017, 175, 828–847. [Google Scholar] [CrossRef]
- Rey, E.; Abrouk, M.; Keeble-Gagnere, G.; Karafiatova, M.; Vrana, J.; Balzergue, S.; Soubigou-Taconnat, L.; Brunaud, V.; Martin-Magniette, M.L.; Endo, T.R.; et al. Transcriptome reprogramming due to the introduction of a barley telosome into bread wheat affects more barley genes than wheat. Plant Biotechnol. J. 2018, 16, 1767–1777. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; Haerum, B.K.; Clark, A.G. Regulatory changes underlying expression differences within and between Drosophila species. Nat. Genet. 2008, 40, 346–350. [Google Scholar] [CrossRef]
- Tirosh, I.; Reikhav, S.; Levy, A.A.; Barkai, N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 2009, 324, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.G.; Ruvinsky, I.; Gilad, Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012, 13, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Barbosa-Morais, N.L.; Schmidt, D.; Conboy, C.M.; Vanes, L.; Tybulewicz, V.L.; Fisher, E.M.; Tavare, S.; Odom, D.T. Species-specific transcription in mice carrying human chromosome 21. Science 2008, 322, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Kazuki, Y.; Gao, F.J.; Yamakawa, M.; Hirabayashi, M.; Kazuki, K.; Kajitani, N.; Miyagawa-Tomita, S.; Abe, S.; Sanbo, M.; Hara, H.; et al. A transchromosomic rat model with human chromosome 21 shows robust Down syndrome features. Am. J. Hum. Genet. 2022, 109, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yu, J.; Li, H.; Huang, W.; Xu, L.; Zhao, Y.; Zhang, T.; Xu, W.; Jiang, J.; Su, Z.; et al. Transcriptional and epigenetic adaptation of maize chromosomes in oat-maize addition lines. Nucleic Acids Res. 2018, 46, 5012–5028. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Wu, X.M.; Bhat, S.R. Brassica and its close allies: Cytogenetics and evolution. Plant Breed. Rev. 2011, 35, 19–84. [Google Scholar]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.C.; Falentin, C.; Genete, M.; Berrabah, W.; Chevre, A.M.; Delourme, R.; et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.K.; et al. A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 2020, 6, 929–941. [Google Scholar] [CrossRef]
- Song, X.; Wei, Y.; Xiao, D.; Gong, K.; Sun, P.; Ren, Y.; Yuan, J.; Wu, T.; Yang, Q.; Li, X.; et al. Brassica carinata genome characterization clarifies U’s triangle model of evolution and polyploidy in Brassica. Plant Physiol. 2021, 186, 388–406. [Google Scholar] [CrossRef]
- Yim, W.C.; Swain, M.L.; Ma, D.; An, H.; Bird, K.A.; Curdie, D.D.; Wang, S.; Ham, H.D.; Luzuriaga-Neira, A.; Kirkwood, J.S.; et al. The final piece of the Triangle of U: Evolution of the tetraploid Brassica carinata genome. Plant Cell 2022, 34, 4141–4172. [Google Scholar] [CrossRef]
- Tan, C.; Cui, C.; Xiang, Y.; Ge, X.; Li, Z. Development of Brassica oleracea-nigra monosomic alien addition lines: Genotypic, cytological and morphological analyses. Theor. Appl. Genet. 2017, 130, 2491–2504. [Google Scholar] [CrossRef]
- He, Z.; Ji, R.; Havlickova, L.; Wang, L.; Li, Y.; Lee, H.T.; Song, J.; Koh, C.; Yang, J.; Zhang, M.; et al. Genome structural evolution in Brassica crops. Nat. Plants 2021, 7, 757–765. [Google Scholar] [CrossRef]
- Nagaharu, U.J. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Shao, Y.; Du, K.; Ran, L.; Fang, X.; Wang, Y. Use of digital gene expression to discriminate gene expression differences in early generations of resynthesized Brassica napus and its diploid progenitors. BMC Genom. 2013, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Shao, Y.; Pan, Q.; Ge, X.; Li, Z. Genome-wide gene expression perturbation induced by loss of C2 chromosome in allotetraploid Brassica napus L. Front. Plant Sci. 2015, 6, 763. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef]
- Schiavoni, F.; Zuazua-Villar, P.; Roumeliotis, T.I.; Benstead-Hume, G.; Pardo, M.; Pearl, F.M.G.; Choudhary, J.S.; Downs, J.A. Aneuploidy tolerance caused by BRG1 loss allows chromosome gains and recovery of fitness. Nat. Commun. 2022, 13, 1731. [Google Scholar] [CrossRef]
- Henry, I.M.; Dilkes, B.P.; Comai, L. Molecular karyotyping and aneuploidy detection in Arabidopsis thaliana using quantitative fluorescent polymerase chain reaction. Plant J. 2006, 48, 307–319. [Google Scholar] [CrossRef]
- Prestel, M.; Feller, C.; Becker, P. Dosage compensation and the global re-balancing of aneuploid genomes. Genome Biol. 2010, 11, 216. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Torres, E.M.; Dunham, M.J.; Amon, A. Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. USA 2012, 109, 12644–12649. [Google Scholar] [CrossRef]
- Ahlfors, H.; Anyanwu, N.; Pakanavicius, E.; Dinischiotu, N.; Lana-Elola, E.; Watson-Scales, S.; Tosh, J.; Wiseman, F.; Briscoe, J.; Page, K.; et al. Gene expression dysregulation domains are not a specific feature of Down syndrome. Nat. Commun. 2019, 10, 2489. [Google Scholar] [CrossRef]
- Cui, C.; Ge, X.; Gautam, M.; Kang, L.; Li, Z. Cytoplasmic and genomic effects on meiotic pairing in Brassica hybrids and allotetraploids from pair crosses of three cultivated diploids. Genetics 2012, 191, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Schelfhout, C.J.; Snowdon, R.; Cowling, W.A.; Wroth, J.M. A PCR based B-genome-specific marker in Brassica species. Theor. Appl. Genet. 2004, 109, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, C.; Cui, C.; Ge, X.; Li, Z. Distinct subgenome stabilities in synthesized Brassica allohexaploids. Theor. Appl. Genet. 2016, 129, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Liu, H.L.; Luo, P. Production and cytogenetics of intergeneric hybrids between Brassica napus and Orychophragmus violaceus. Theor. Appl. Genet. 1995, 91, 131–136. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jacob, A.; Lancaster, J.; Buhler, J.; Harris, B.; Chamberlain, R.D. Mercury BLASTP: Accelerating Protein Sequence Alignment. ACM Trans. Reconfigurable Technol. Syst. 2008, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Up Regulated Genes | Ratio (%) | Down Regulated Genes | Ratio (%) | Total | Ratio (%) |
|---|---|---|---|---|---|---|
| CCB1 vs. CC | 861 | 41.51 | 1213 ** | 58.49 | 2074 A | 3.12 |
| CCB4 vs. CC | 547 | 38.66 | 868 ** | 61.34 | 1415 A | 2.12 |
| CCB5 vs. CC | 559 | 41.47 | 789 ** | 58.53 | 1348 A | 1.99 |
| CCB6 vs. CC | 1336 | 46.10 | 1562 ** | 53.90 | 2898 B | 4.32 |
| CCB8 vs. CC | 6167 | 50.78 | 5977 | 49.22 | 12144 C | 17.88 |
| CCn1 vs. CC | 354 | 52.68 | 318 | 47.32 | 672 a | 1.12 |
| CCn4 vs. CC | 165 ** | 58.72 | 116 | 41.28 | 281 a, b | 0.47 |
| CCn5 vs. CC | 154 | 49.52 | 157 | 50.48 | 311 a | 0.52 |
| CCn6 vs. CC | 424 ** | 64.44 | 234 | 35.56 | 658 b | 1.09 |
| CCn8 vs. CC | 2642 ** | 54.75 | 2184 | 45.25 | 4826 a | 8.02 |
| Comparisons | Trans-Effects | Cis-Effects | ||||
|---|---|---|---|---|---|---|
| Up- (%) | Down- (%) | Total (%) | Up- (%) | Down- (%) | Total (%) | |
| CCB1 vs. CC | 694 (45.24) | 840 (54.76) ** | 1534 (2.55) A | 167 (30.93) | 373 (69.07) ** | 540 (8.75) A |
| CCB4 vs. CC | 379 (46.11) | 443 (53.89) * | 822 (1.37) A | 168 (28.33) | 425 (71.67) ** | 593 (9.05) A |
| CCB5 vs. CC | 332 (49.70) | 336 (50.30) | 668 (1.11) A, B | 227 (33.38) | 453 (66.62) ** | 680 (8.92) A |
| CCB6 vs. CC | 1121 (49.54) | 1142 (50.46) | 2263 (3.76) A, B | 215 (33.86) | 420 (66.14) ** | 635 (9.24) A |
| CCB8 vs. CC | 5522 (52.10) ** | 5077 (47.90) | 10599 (17.60) B | 645 (41.75) | 900 (58.25) ** | 1545 (20.03) B |
| Gene ID | Up/Down | Putative Orthologs in A. thaliana | Gene Functions |
|---|---|---|---|
| BolC1t04413H | Down | AT3G19640 | Magnesium ion transmembrane transporter activity, metal ion transmembrane transporter activity |
| BolC1t05746H | Down | AT1G60720 | RNA-directed DNA polymerase (reverse transcriptase)-related family protein |
| BolC2t09633H | Down | AT3G32904 | Unknown protein |
| BolC2t11658H | Down | AT1G10000 | Ribonuclease H activity, nucleic acid binding |
| BolC3t13980H | Up | AT2G32260 | Phosphorylcholine cytidylyltransferase |
| BolC3t14199H | Up/Down (B8/n8) | AT5G53160 | Regulatory components of ABA receptor 3 |
| BolC3t15755H | Down | AT4G09340 | SPla/RYanodine receptor (SPRY) domain-containing protein |
| BolC4t23401H | Down | AT2G31470 | F-box and associated interaction domain-containing protein |
| BolC4t24470H | Down | AT1G63670 | Protein of unknown function (DUF3741) |
| BolC6t37425H | Up | AT2G32260 | Phosphorylcholine cytidylyltransferase |
| BolC6t39384H | Up | AT1G33590 | Leucine-rich repeat (LRR) family protein |
| BolC6t39403H | Up/Down (B8) | AT2G33150 | Peroxisomal 3-ketoacyl-CoA thiolase 3 |
| MAALs | Up in CC | Down in CC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Unexpressed in BB (%) | Down in BB (%) | Up in BB (%) | Unchanged in BB (%) | Total (%) | Unexpressed in BB (%) | Down in BB (%) | Up in BB (%) | Unchanged in BB (%) | Total (%) | |
| CCB1 | 13 (22.42) | 24 (41.38) | 6 (10.34) | 15 (25.86) | 58 (2.04) | 16 (20.25) | 36 (45.57) | 4 (5.06) | 23 (29.12) | 79 (2.78) |
| CCB4 | 12 (27.91) | 19 (44.18) | 1 (2.33) | 11 (25.58) | 43 (1.47) | 28 (28.28) | 46 (46.47) | 1 (1.01) | 24 (24.24) | 99 (3.38) |
| CCB5 | 10 (21.28) | 22 (46.81) | 6 (12.77) | 9 (19.14) | 47 (1.42) | 34 (36.96) | 31 (33.70) | 7 (7.61) | 20 (21.73) | 92 (2.78) |
| CCB6 | 41 (27.52) | 56 (37.58) | 11 (7.38) | 41 (27.52) | 149 (4.60) | 44 (34.11) | 35 (27.13) | 10 (7.75) | 40 (31.01) | 129 (3.98) |
| CCB8 | 75 (12.25) | 95 (15.52) | 181 (29.58) | 261 (42.65) | 612 (18.21) | 40 (10.15) | 300 (76.14) | 10 (2.54) | 44 (11.17) | 394 (11.73) |
| Comparisons | Low (1 < TPM < 10) | Medium (10 < TPM < 100) | High (TPM > 100) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Up | Down | Total | Ratio | Up | Down | Total | Ratio | Up | Down | Total | Ratio | |
| CCB1 vs. CC | 309 | 439 ** | 748 | 5.69% | 123 | 339 ** | 462 | 4.50% | 13 | 62 ** | 75 | 5.60% |
| CCn1 vs. CC | 144 | 176 | 320 | 2.43% | 46 | 127 ** | 173 | 1.69% | 5 | 15 | 20 | 1.49% |
| CCB4 vs. CC | 140 | 268 ** | 408 | 3.10% | 55 | 160 ** | 215 | 2.10% | 6 | 15 | 21 | 1.57% |
| CCn4 vs. CC | 47 | 64 | 111 | 0.84% | 15 | 49 ** | 64 | 0.62% | 1 | 3 | 4 | 0.30% |
| CCB5 vs. CC | 122 | 180 ** | 302 | 2.30% | 47 | 130 ** | 177 | 1.72% | 4 | 22 ** | 26 | 1.94% |
| CCn5 vs. CC | 20 | 88 ** | 138 | 1.05% | 30 | 59 | 89 | 0.87% | 1 | 10 | 11 | 0.82% |
| CCB6 vs. CC | 469 | 628 ** | 1097 | 8.34% | 142 | 395 ** | 537 | 5.23% | 10 | 98 ** | 108 | 8.06% |
| CCn6 vs. CC | 171 | 128 | 299 | 2.27% | 73 | 91 | 164 | 1.60% | 8 | 15 | 23 | 1.72% |
| CCB8 vs. CC | 2245 | 2098 | 4343 | 33.04% | 1046 | 2465 ** | 3511 | 34.21% | 147 | 514 ** | 661 | 49.33% |
| CCn8 vs. CC | 1171 ** | 890 | 2061 | 15.68% | 732 | 1078 ** | 1810 | 17.64% | 93 | 216 ** | 309 | 23.06% |
| Comparisons | Down in MAALs | Up in MAALs | ||||||
|---|---|---|---|---|---|---|---|---|
| Up- (%) | Down- (%) | Unchanged- (%) | Total (%) | Up- (%) | Down- (%) | Unchanged- (%) | Total (%) | |
| CCn1 vs. CC | 3 (0.36) | 269 (32.02) | 568 (67.62) | 840 (100) | 284 (40.92) | 4 (0.58) | 406 (58.50) | 694 (100) |
| CCn4 vs. CC | 1 (0.22) | 172 (38.83) | 270 (60.95) | 443 (100) | 239 (63.06) | 0 (0) | 140 (36.94) | 379 (100) |
| CCn5 vs. CC | 2 (0.60) | 196 (59.04) | 134 (40.36) | 332 (100) | 237 (70.54) | 0 (0) | 99 (29.46) | 336 (100) |
| CCn6 vs. CC | 8 (0.71) | 383 (34.17) | 730 (65.12) | 1121 (100) | 478 (41.86) | 5 (0.44) | 659 (57.70) | 1142 (100) |
| CCn8 vs. CC | 158 (3.11) | 1802 (35.49) | 3117 (61.40) | 5077 (100) | 2048 (37.09) | 78 (1.41) | 3396 (61.50) | 5522 (100) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, P.; Ge, X.; Li, Z. Transcriptional Interactions of Single B-Subgenome Chromosome with C-Subgenome in B. oleracea-nigra Additional Lines. Plants 2023, 12, 2029. https://doi.org/10.3390/plants12102029
Zeng P, Ge X, Li Z. Transcriptional Interactions of Single B-Subgenome Chromosome with C-Subgenome in B. oleracea-nigra Additional Lines. Plants. 2023; 12(10):2029. https://doi.org/10.3390/plants12102029
Chicago/Turabian StyleZeng, Pan, Xianhong Ge, and Zaiyun Li. 2023. "Transcriptional Interactions of Single B-Subgenome Chromosome with C-Subgenome in B. oleracea-nigra Additional Lines" Plants 12, no. 10: 2029. https://doi.org/10.3390/plants12102029
APA StyleZeng, P., Ge, X., & Li, Z. (2023). Transcriptional Interactions of Single B-Subgenome Chromosome with C-Subgenome in B. oleracea-nigra Additional Lines. Plants, 12(10), 2029. https://doi.org/10.3390/plants12102029