
Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae


Abstract
:1. Introduction
2. Results
2.1. Genetic Variation of ITS2 Sequences in Pinaceae
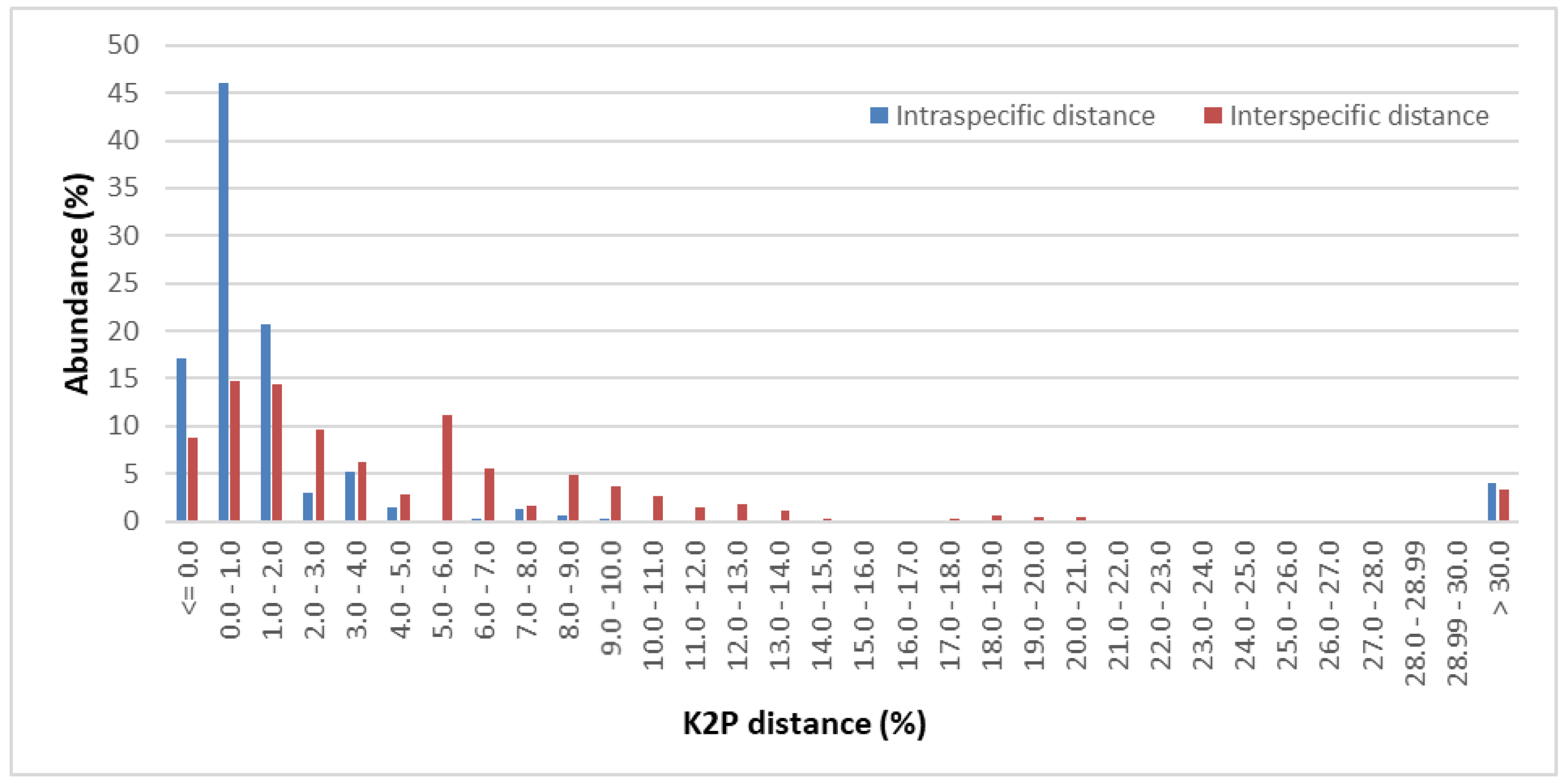
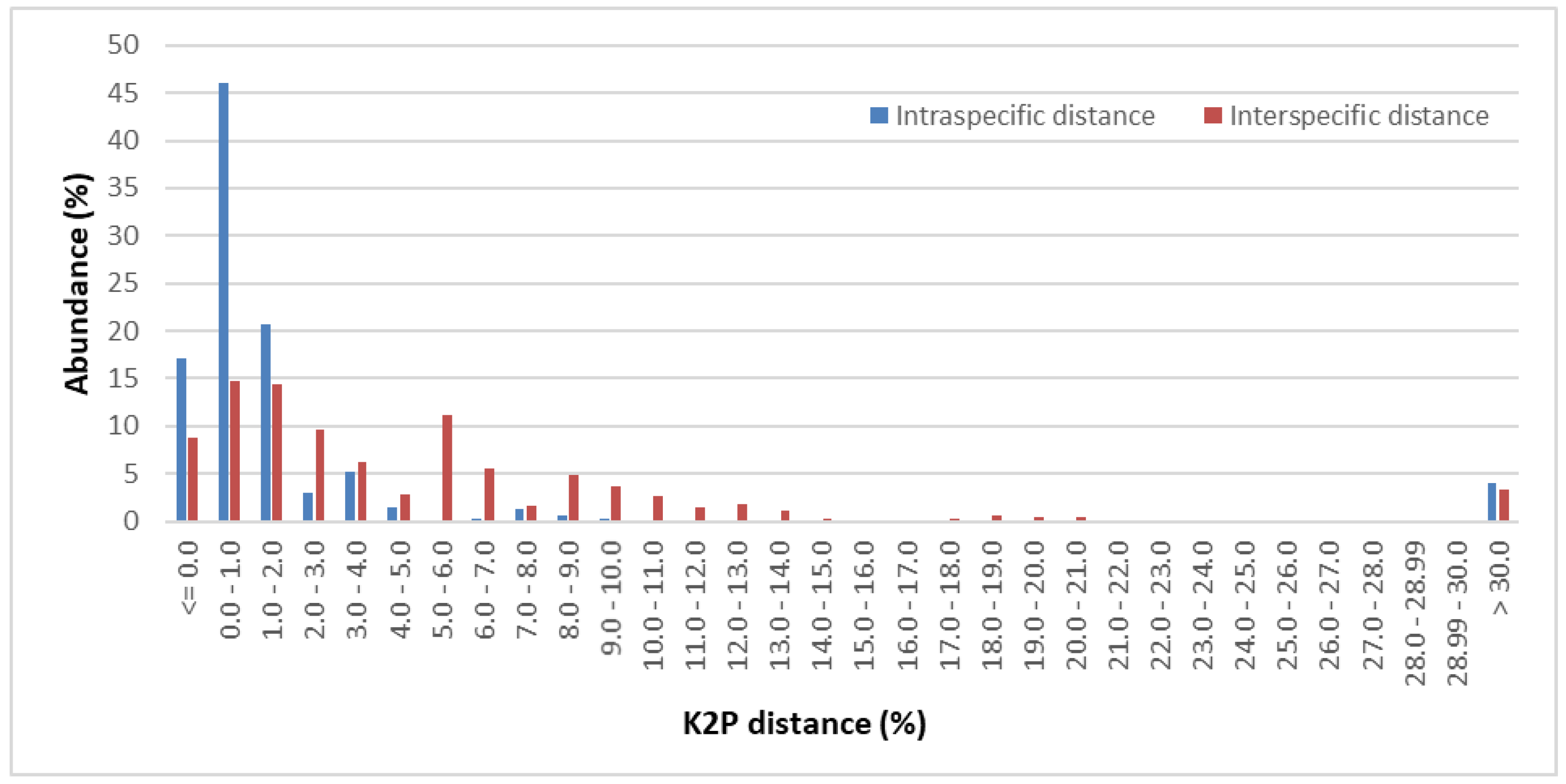
2.2. Genetic Divergence within and between Pinaceae Taxa
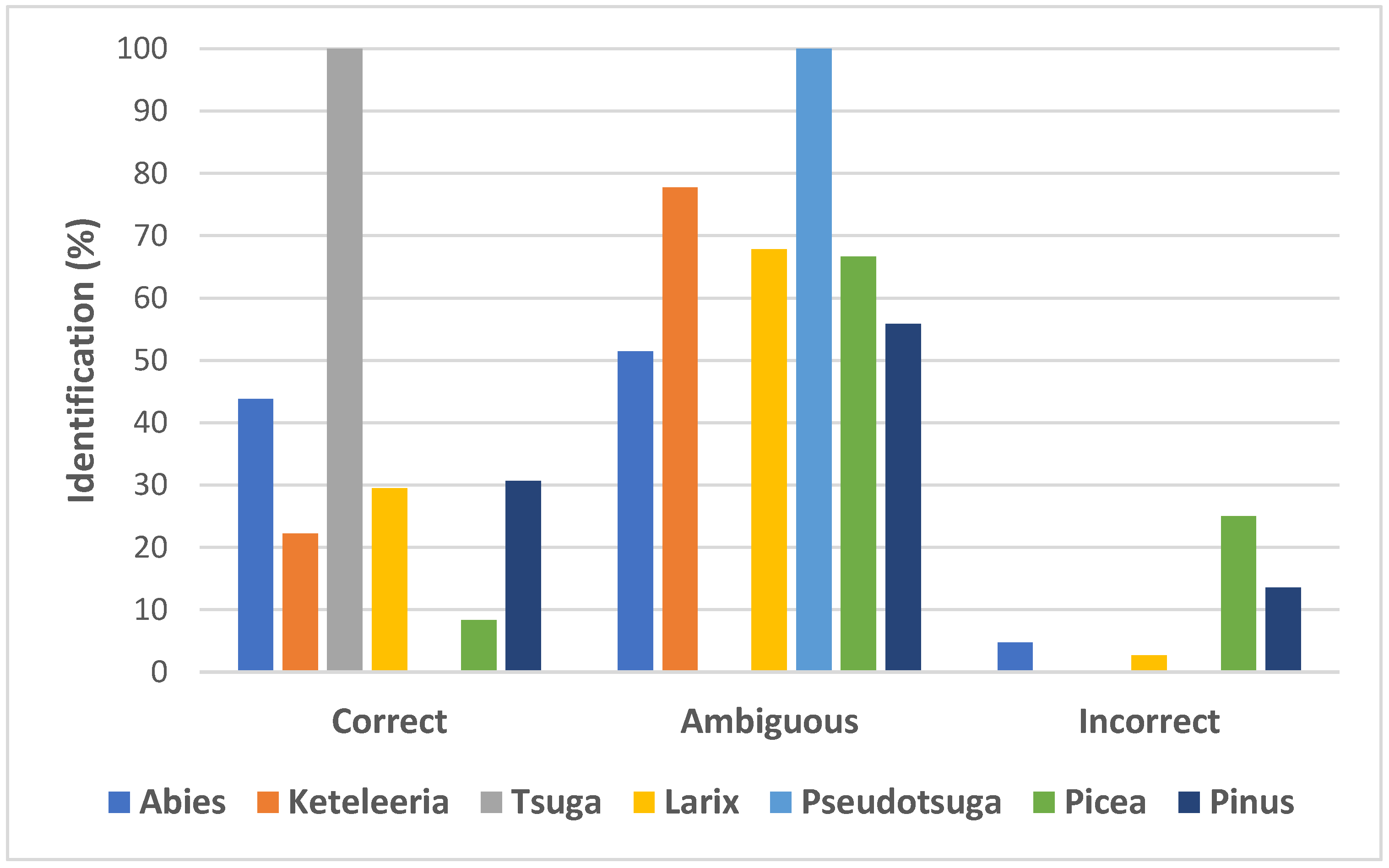
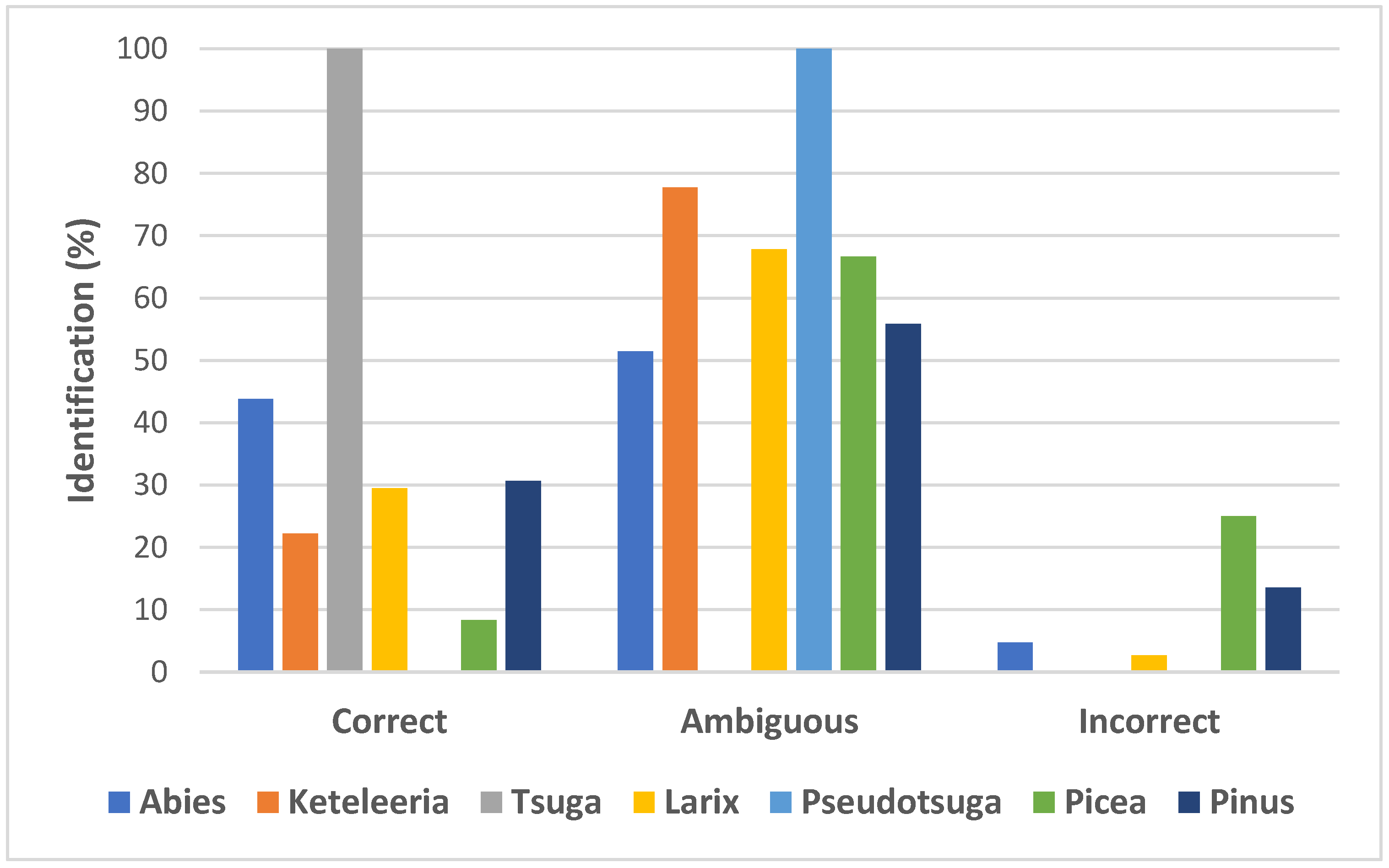
2.3. Rates of Sequences Identification and Taxa Discrimination among Pinaceae
2.4. Phylogenetic Inference
3. Discussion
4. Materials and Methods
4.1. Sampling and Plant Materials
4.2. DNA Extraction and Next-Generation Sequencing
4.3. Data Validation
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liston, A.; Vining, T.F.; Campbell, C.S.; Gernandt, D.S.; Piñero, D. Molecular phylogeny of pinaceae and pinus. Acta Hortic. 2003, 615, 107–114. [Google Scholar] [CrossRef]
- Farjon, A. A Natural History of Conifers; Timber Press: Portland, OR, USA, 2008. [Google Scholar]
- Le Maitre, D.C. Pines in cultivation: A global view. In Ecology and Biogeography of Pinus; David, M.R., Ed.; Cambridge University Press: Cambridge, UK, 1998; pp. 407–431. [Google Scholar]
- Leduc, C.; Coonishish, J.; Haddad, P.; Cuerrier, A. Plants used by the cree nation of eeyou istchee (Quebec, Canada) for the treatment of diabetes: A novel approach in quantitative ethnobotany. J. Ethnopharmacol. 2006, 105, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Yasin, M.; Hussain Janbaz, K.; Imran, I.; Gilani, A.U.H.; Bashir, S. Pharmacological Studies on the antispasmodic, bronchodilator and anti-platelet activities of abies webbiana. Phyther. Res. 2014, 28, 1182–1187. [Google Scholar] [CrossRef]
- Kizilarslan, Ç.; Sevg, E. Ethnobotanical uses of genus pinus, L. (Pinaceae) in Turkey. Indian J. Tradit. Knowl. 2013, 12, 209–220. [Google Scholar]
- Hussain, K.; Nisar, M.F.; Majeed, A.; Nawaz, K.; Bhatti, K.H. Ethnomedicinal survey for important plants of Jalalpur Jattan, District Gujrat, Punjab, Pakistan. Ethnobot. Leafl. 2010, 14, 807–825. [Google Scholar]
- Kaushik, P.; Lal, S.; Rana, A.C.; Kaushik, D. GC-MS analysis of bioactive constituents of pinus roxburghii sarg. (Pinaceae) from Northern India. Res. J. Phytochem. 2014, 8, 42–46. [Google Scholar] [CrossRef]
- Celiński, K.; Kijak, H.; Wojnicka-Półtorak, A.; Buczkowska-Chmielewska, K.; Sokołowska, J.; Chudzińska, E. Effectiveness of the DNA barcoding approach for closely related conifers discrimination: A case study of the pinus mugo complex. Comptes Rendus-Biol. 2017, 340, 339–348. [Google Scholar] [CrossRef]
- Celiński, K.; Bonikowski, R.; Wojnicka-Półtorak, A.; Chudzińska, E.; Maliński, T. Volatiles as chemosystematic markers for distinguishing closely related species within the pinus mugo complex. Chem. Biodivers. 2015, 12, 1208–1213. [Google Scholar] [CrossRef]
- Bonikowski, R.; Celiński, K.; Wojnicka-Półtorak, A.; Maliński, T. Composition of essential oils isolated from the needles of Pinus Uncinata and P. Uliginosa Grown in Poland. Nat. Prod. Commun. 2015, 10, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Celiński, K.; Kijak, H.; Barylski, J.; Grabsztunowicz, M.; Wojnicka-Półtorak, A.; Chudzińska, E. Characterization of the complete chloroplast genome of pinus uliginosa (neumann) from the pinus mugo complex. Conserv. Genet. Resour. 2017, 9, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Celiński, K.; Chudzińska, E.; Gmur, A.; Piosik, Ł.; Wojnicka-Półtorak, A. Cytological characterization of three closely related pines—Pinus Mugo, P. Uliginosa and P. × Rhaetica from the Pinus Mugo complex (Pinaceae). Biologia 2019, 74, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Wachowiak, W.; Celiński, K.; Prus-Głowacki, W. Evidence of natural reciprocal hybridisation between Pinus Uliginosa and P. Sylvestris in the sympatric population of the species. Flora Morphol. Distrib. Funct. Ecol. Plants 2005, 200, 563–568. [Google Scholar] [CrossRef]
- Sokołowska, J.; Fuchs, H.; Celiński, K. New insight into taxonomy of european mountain pines, pinus mugo complex, based on complete chloroplast genomes sequencing. Plants 2021, 10, 1331. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Wang, X.C.; Liu, C.; Huang, L.; Bengtsson-Palme, J.; Chen, H.; Zhang, J.H.; Cai, D.; Li, J.Q. ITS1: A DNA barcode better than ITS2 in eukaryotes? Mol. Ecol. Resour. 2015, 15, 573–586. [Google Scholar] [CrossRef]
- Ran, J.H.; Wang, P.P.; Zhao, H.J.; Wang, X.Q. A test of seven candidate barcode regions from the Plastome in Picea (Pinaceae). J. Integr. Plant Biol. 2010, 52, 1109–1126. [Google Scholar] [CrossRef]
- Little, D.P.; Knopf, P.; Schulz, C. DNA barcode identification of Podocarpaceae—The second largest conifer family. PLoS ONE 2013, 8, e81008. [Google Scholar] [CrossRef] [Green Version]
- Maia, V.H.; da Mata, C.S.; Franco, L.O.; Cardoso, M.A.; Cardoso, S.R.S.; Hemerly, A.S.; Ferreira, P.C.G. DNA barcoding bromeliaceae: Achievements and pitfalls. PLoS ONE 2012, 7, e29877. [Google Scholar] [CrossRef] [Green Version]
- LV, Y.N.; YANG, C.Y.; SHI, L.C.; ZHANG, Z.L.; XU, A.S.; ZHANG, L.X.; LI, X.L.; LI, H.T. Identification of Medicinal plants within the apocynaceae family using ITS2 and PsbA-TrnH barcodes. Chin. J. Nat. Med. 2020, 18, 594–605. [Google Scholar] [CrossRef]
- Gao, T.; Yao, H.; Song, J.; Liu, C.; Zhu, Y.; Ma, X.; Pang, X.; Xu, H.; Chen, S. Identification of Medicinal plants in the family fabaceae using a potential DNA barcode ITS2. J. Ethnopharmacol. 2010, 130, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zeng, X.; Yang, D.; Chu, G.; Yuan, Z.; Chen, S. Applying DNA barcodes for identification of plant species in the family araliaceae. Gene 2012, 499, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jiang, M.; Shi, Y.; Jiao, K.; Shen, C.; Lu, J.; Ying, Q.; Wang, H. Application of the ribosomal DNA ITS2 region of physalis (Solanaceae): DNA barcoding and phylogenetic study. Front. Plant Sci. 2016, 7, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Song, J.; Liu, C.; Luo, K.; Han, J.; Li, Y.; Pang, X.; Xu, H.; Zhu, Y.; Xiao, P.; et al. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS ONE 2010, 5, e13102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef]
- Coleman, A.W. ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet. 2003, 19, 370–375. [Google Scholar] [CrossRef]
- Schultz, J.; Maisel, S.; Gerlach, D.; Müller, T.; Wolf, M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 2005, 11, 361–364. [Google Scholar] [CrossRef] [Green Version]
- Miao, M.; Warren, A.; Song, W.; Wang, S.; Shang, H.; Chen, Z. Analysis of the internal transcribed spacer 2 (ITS2) region of scuticociliates and related taxa (ciliophora, oligohymenophorea) to infer their evolution and phylogeny. Protist 2008, 159, 519–533. [Google Scholar] [CrossRef]
- Coleman, A.W. Pan-eukaryote ITS2 homologies revealed by RNA secondary structure. Nucleic Acids Res. 2007, 35, 3322–3329. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.; Wolf, M. ITS2 sequence-structure analysis in phylogenetics: A how-to manual for molecular systematics. Mol. Phylogenetics Evol. 2009, 52, 520–523. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Song, J.; Zhu, Y.; Xie, C.; Chen, S. Using DNA barcoding to identify species within euphorbiaceae. Planta Med. 2010, 76, 1784–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Yao, H.; Song, J.; Zhu, Y.; Liu, C.; Chen, S. Evaluating the feasibility of using candidate DNA barcodes in discriminating species of the large Asteraceae family. BMC Evol. Biol. 2010, 10, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- China Plant BOL Group; Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; et al. Comparative analysis of a large dataset indicates that Internal Transcribed Spacer (ITS) Should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, B.G.; Sanderson, M.J.; Porter, J.M.; Wojciechowski, M.F.; Campbell, C.S.; Donoghue, M.J. The ITS Region of nuclear ribosomal DNA: A valuable source of evidence on angiosperm phylogeny. Ann. Missouri Bot. Gard. 1995, 85, 247–277. [Google Scholar] [CrossRef]
- Liston, A.; Robinson, W.A.; Oliphant, J.M.; Alvarez-Buylla, E.R. Length variation in the nuclear ribosomal DNA internal transcribed spacer region of non-flowering seed plants. Syst. Bot. 1996, 21, 109–120. [Google Scholar] [CrossRef]
- Marrocco, R.; Gelati, M.T.; Maggini, F. Nucleotide sequence of the internal transcribed spacers and 5.8 s region of ribosomal DNA in Phus pinea L. Mitochondrial DNA 1996, 6, 175–177. [Google Scholar] [CrossRef]
- Vining, T.F.; Campbell, C.S. Phylogenetic signal in sequence repeats within nuclear ribosomal DNA internal transcribed spacer 1 in Tsuga. Am. J. Bot. 1997, 84, 702. [Google Scholar]
- Gernandt, D.S.; Liston, A. Internal transcribed spacer region evolution in Larix and Pseudotsuga (Pinaceae). Am. J. Bot. 1998, 86, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Maggini, F.; Frediani, M.; Gelati, M.T. Nucleotide sequence of the internal transcribed spacers of ribosomal DNA in Picea Abies Karst. Mitochondrial DNA 2000, 11, 87–89. [Google Scholar] [CrossRef]
- Han, J.; Zhu, Y.; Chen, X.; Liao, B.; Yao, H.; Song, J.; Chen, S.; Meng, F. The short ITS2 sequence serves as an efficient taxonomic sequence tag in comparison with the full-length ITS. Biomed Res. Int. 2013, 2013, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazekas, A.J.; Burgess, K.S.; Kesanakurti, P.R.; Graham, S.W.; Newmaster, S.G.; Husband, B.C.; Percy, D.M.; Hajibabaei, M.; Barrett, S.C.H. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE 2008, 3, e2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, W.J.; Erickson, D.L. A Two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-PsbA spacer region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newmaster, S.G.; Fazekas, A.J.; Ragupathy, S. DNA barcoding in land plants: Evaluation of RbcL in a multigene tiered approach. Can. J. Bot. 2006, 84, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, E. Taxonomy. Wanted: A barcode for plants. Science 2007, 318, 190–191. [Google Scholar] [CrossRef]
- Chase, M.W.; Cowan, R.S.; Hollingsworth, P.M.; Van Den Berg, C.; Madriñán, S.; Petersen, G.; Seberg, O.; Jørgsensen, T.; Cameron, K.M.; Carine, M.; et al. A Proposal for a standardised protocol to barcode all land plants. Taxon 2007, 56, 295–299. [Google Scholar] [CrossRef]
- Lahaye, R.; Van Der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.S.; Ayres, K.L.; Toomey, N.; Haider, N.; Stahl, J.V.A.; Kelly, L.J.; Wikström, N.; Hollingsworth, P.M.; Duff, R.J.; Hoot, S.B.; et al. Selection of candidate coding DNA barcoding regions for use on land plants. Bot. J. Linn. Soc. 2009, 159, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Starr, J.R.; Naczi, R.F.C.; Chouinard, B.N. Plant DNA barcodes and species resolution in sedges (Carex, Cyperaceae). Mol. Ecol. Resour. 2009, 9 (Suppl. S1), 151–163. [Google Scholar] [CrossRef]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA. 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newmaster, S.G.; Ragupathy, S. Esting plant barcoding in a sister species complex of pantropical acacia (Mimosoideae, Fabaceae). Mol. Ecol. Resour. 2009, 9, 172–180. [Google Scholar]
- Pang, X.; Liu, C.; Shi, L.; Liu, R.; Liang, D.; Li, H.; Cherny, S.S.; Chen, S. Utility of the TrnH-PsbA intergenic spacer region and its combinations as plant DNA barcodes: A meta-analysis. PLoS ONE 2012, 7, e48833. [Google Scholar] [CrossRef]
- Bolson, M.; De Camargo Smidt, E.; Brotto, M.L.; Silva-Pereira, V. ITS and TrnH-PsbA as efficient DNA barcodes to identify threatened commercial woody angiosperms from Southern Brazilian atlantic rainforests. PLoS ONE 2015, 10, e0143049. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [Green Version]
- Olsson, S.; Grivet, D.; Cid Vian, J. Species-diagnostic markers in the genus pinus: Evaluation of the chloroplast regions Matk and Ycf1. For. Syst. 2018, 27, e016. [Google Scholar] [CrossRef] [Green Version]
- Zeb, U.; Dong, W.L.; Zhang, T.T.; Wang, R.N.; Shahzad, K.; Ma, X.F.; Li, Z.H. Comparative plastid genomics of pinus species: Insights into sequence variations and phylogenetic relationships. J. Syst. Evol. 2020, 58, 118–132. [Google Scholar] [CrossRef]
- Kormutak, A.; Galgoci, M.; Bolecek, P.; Gomory, D.; Libantova, J. Reinforced evidence on partial compatibility between pinus sylvestris and pinus mugo and on maternal inheritance of chloroplast DNA in the Pinus Mugo × Pinus Sylvestris cross. Silvae Genet. 2020, 69, 108–115. [Google Scholar] [CrossRef]
- Luo, K.; Chen, S.L.; Chen, K.L.; Song, J.Y.; Yao, H.; Ma, X.; Zhu, Y.J.; Pang, X.H.; Yu, H.; Li, X.W.; et al. Assessment of candidate plant DNA barcodes using the rutaceae family. Sci. China Life Sci. 2010, 53, 701–708. [Google Scholar] [CrossRef]
- Pang, X.; Song, J.; Zhu, Y.; Xu, H.; Huang, L.; Chen, S. Applying plant DNA barcodes for rosaceae species identification. Cladistics 2011, 27, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Q.; Qu, Y.Q.; Yao, H.; Zhang, Y.M.; Yan, Q.Q.; Hang, Y.Y. Applying DNA barcodes for identification of economically important species in Brassicaceae. Genet. Mol. Res. 2015, 14, 15050–15061. [Google Scholar] [CrossRef] [PubMed]
- Koetschan, C.; Förster, F.; Keller, A.; Schleicher, T.; Ruderisch, B.; Schwarz, R.; Müller, T.; Wolf, M.; Schultz, J. The ITS2 database III sequences and structures for phylogeny. Nucleic Acids Res. 2009, 38 (Suppl. S1), 275–279. [Google Scholar] [CrossRef] [PubMed]
- Selig, C.; Wolf, M.; Müller, T.; Dandekar, T.; Schultz, J. The ITS2 database II: Homology modelling RNA Structure for molecular systematics. Nucleic Acids Res. 2008, 36 (Suppl. S1), 377–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Müller, T.; Achtziger, M.; Seibel, P.N.; Dandekar, T.; Wolf, M. The internal transcribed spacer 2 database—A web server for (not only) low level phylogenetic analyses. Nucleic Acids Res. 2006, 34, 704–707. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kimura, M. A Simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef] [Green Version]
- Meier, R.; Zhang, G.; Ali, F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst. Biol. 2008, 57, 809–813. [Google Scholar] [CrossRef]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Taxonomic Group/Level | Name | AL | CS | VS | PIS | SS | OMD |
|---|---|---|---|---|---|---|---|
| Species complex | P. mugo complex | 243 | 243 (100.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0.000 |
| Genera | Abies | 246 | 169 (68.70) | 75 (30.49) | 27 (10.98) | 48 (19.51) | 0.016 |
| Keteleeria | 249 | 118 (47.39) | 122 (48.99) | 0 (0.00) | 121 (48.5) | 0.236 | |
| Tsuga | 247 | 237 (95.95) | 10 (4.05) | 7 (2.83) | 3 (1.21) | 0.026 | |
| Larix | 233 | 172 (73.82) | 61 (26.18) | 15 (6.44) | 46 (19.74) | 0.013 | |
| Pseudotsuga | 232 | 232 (100.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0.000 | |
| Picea | 236 | 232 (98.31) | 4 (1.69) | 3 (1.27) | 1 (0.42) | 0.004 | |
| Pinus | 252 | 70 (27.78) | 177 (70.24) | 73 (28.97) | 101 (40.0) | 0.077 | |
| Subfamily | Abietoideae | 254 | 66 (25.98) | 186 (73.23) | 88 (34.65) | 97 (38.19) | 0.074 |
| Laricoideae | 233 | 148 (63.52) | 85 (36.48) | 48 (20.60) | 37 (15.88) | 0.023 | |
| Pinoideae | 252 | 61 (24.21) | 187 (74.21) | 107 (42.4) | 77 (30.56) | 0.149 | |
| Family | Pinaceae | 270 | 17 (6.30) | 250 (92.59) | 196 (72.5) | 49 (18.15) | 0.342 |
| Taxonomic Group/Level | Name | All Interspecific Distance | Minimum Interspecific Distance | All Intraspecific Distance | Theta | Coalescent Depth |
|---|---|---|---|---|---|---|
| Species complex | P. mugo complex | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 |
| Genera | Abies | 0.0175 ± 0.0006 | 0.0000 ± 0.0000 | 0.0085 ± 0.0012 | 0.0176 ± 0.0039 | 0.0190 ± 0.0046 |
| Keteleeria | 0.1998 ± 0.0973 | 0.0000 ± 0.0000 | 0.1972 ± 0.1919 | 0.1995 ± 0.0250 | 0.2958 ± 0.2874 | |
| Tsuga | 0.0356 ± 0.0119 | 0.0291 ± 0.0109 | 0.0061 ± 0.0061 | 0.0258 ± 00820 | 0.0062±0.0061 | |
| Larix | 0.0137 ± 0.0006 | 0.0000 ± 0.0000 | 0.0067 ± 0.0010 | 0.0130 ± 0.0033 | 0.0271 ± 0.0074 | |
| Pseudotsuga | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | 0.0000 ± 0.0000 | |
| Picea | 0.0048 ± 0.0009 | 0.0000 ± 0.0000 | 0.0017 ± 0.0012 | 0.0043 ± 0.0023 | 0.0024 ± 0.0018 | |
| Pinus | 0.0392 ± 0.0257 | 0.0000 ± 0.0000 | 0.0959 ± 0.0050 | 0.0768 ± 0.0102 | 0.0486 ± 0.0259 | |
| Subfamily | Abietoideae | 0.1089±0.0082 | 0.0000 ± 0.0000 | 0.0370 ± 0.0291 | 0.0665 ± 0.0065 | 0.0562 ± 0.0389 |
| Laricoideae | 0.0436 ± 0.0083 | 0.0000 ± 0.0000 | 0.0063 ± 0.0011 | 0.0222 ± 0.0037 | 0.0249 ± 0.0071 | |
| Pinoideae | 0.1702 ± 0.0059 | 0.0000 ± 0.0000 | 0.0342 ± 0.0229 | 0.1492 ± 0.0157 | 0.0396 ± 0.0210 | |
| Family | Pinaceae | 0.3586 ± 0.0041 | 0.0000 ± 0.0000 | 0.0294 ± 0.0141 | 0.3417 ± 0.0287 | 0.0423 ± 0.0162 |
| Taxonomic Group/Level | Name | Best Match (BM) | Best Close Match (BCM) | |||||
|---|---|---|---|---|---|---|---|---|
| Correct | Incorrect | Ambiguous | Correct | Incorrect | Ambiguous | No Match | ||
| Species complex | P. mugo complex | 0.00 | 0.00 | 100.00 | 0.00 | 0.00 | 100.00 | 0.00 |
| Genera | Abies | 16.19 | 8.57 | 75.24 | 16.19 | 8.57 | 75.24 | 0.00 |
| Keteleeria | 11.11 | 0.00 | 88.89 | 11.10 | 0.00 | 88.89 | 0.00 | |
| Tsuga | 100.00 | 0.00 | 0.00 | 100.00 | 0.00 | 0.00 | 0.00 | |
| Larix | 21.43 | 3.57 | 75.00 | 21.43 | 2.68 | 75.00 | 0.89 | |
| Pseudotsuga | 100.00 | 0.00 | 0.00 | 100.00 | 0.00 | 0.00 | 0.00 | |
| Picea | 20.83 | 0.00 | 79.17 | 20.83 | 0.00 | 79.17 | 0.00 | |
| Pinus | 16.22 | 9.01 | 74.77 | 15.32 | 7.21 | 72.07 | 5.40 | |
| Subfamily | Abietoideae | 17.80 | 8.47 | 73.73 | 17.80 | 7.63 | 73.73 | 0.84 |
| Laricoideae | 23.48 | 3.48 | 73.04 | 23.40 | 2.61 | 75.00 | 0.87 | |
| Pinoideae | 17.04 | 7.40 | 75.50 | 16.30 | 5.93 | 73.33 | 4.44 | |
| Family | Pinaceae | 19.20 | 6.52 | 74.10 | 19.00 | 5.43 | 73.38 | 2.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokołowska, J.; Fuchs, H.; Celiński, K. Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae. Plants 2022, 11, 1078. https://doi.org/10.3390/plants11081078
Sokołowska J, Fuchs H, Celiński K. Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae. Plants. 2022; 11(8):1078. https://doi.org/10.3390/plants11081078
Chicago/Turabian StyleSokołowska, Joanna, Hanna Fuchs, and Konrad Celiński. 2022. "Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae" Plants 11, no. 8: 1078. https://doi.org/10.3390/plants11081078
APA StyleSokołowska, J., Fuchs, H., & Celiński, K. (2022). Assessment of ITS2 Region Relevance for Taxa Discrimination and Phylogenetic Inference among Pinaceae. Plants, 11(8), 1078. https://doi.org/10.3390/plants11081078