
Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L.


 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Morphological Characteristics of L. stelleroides Specimens
2.2. Assembling Sequences
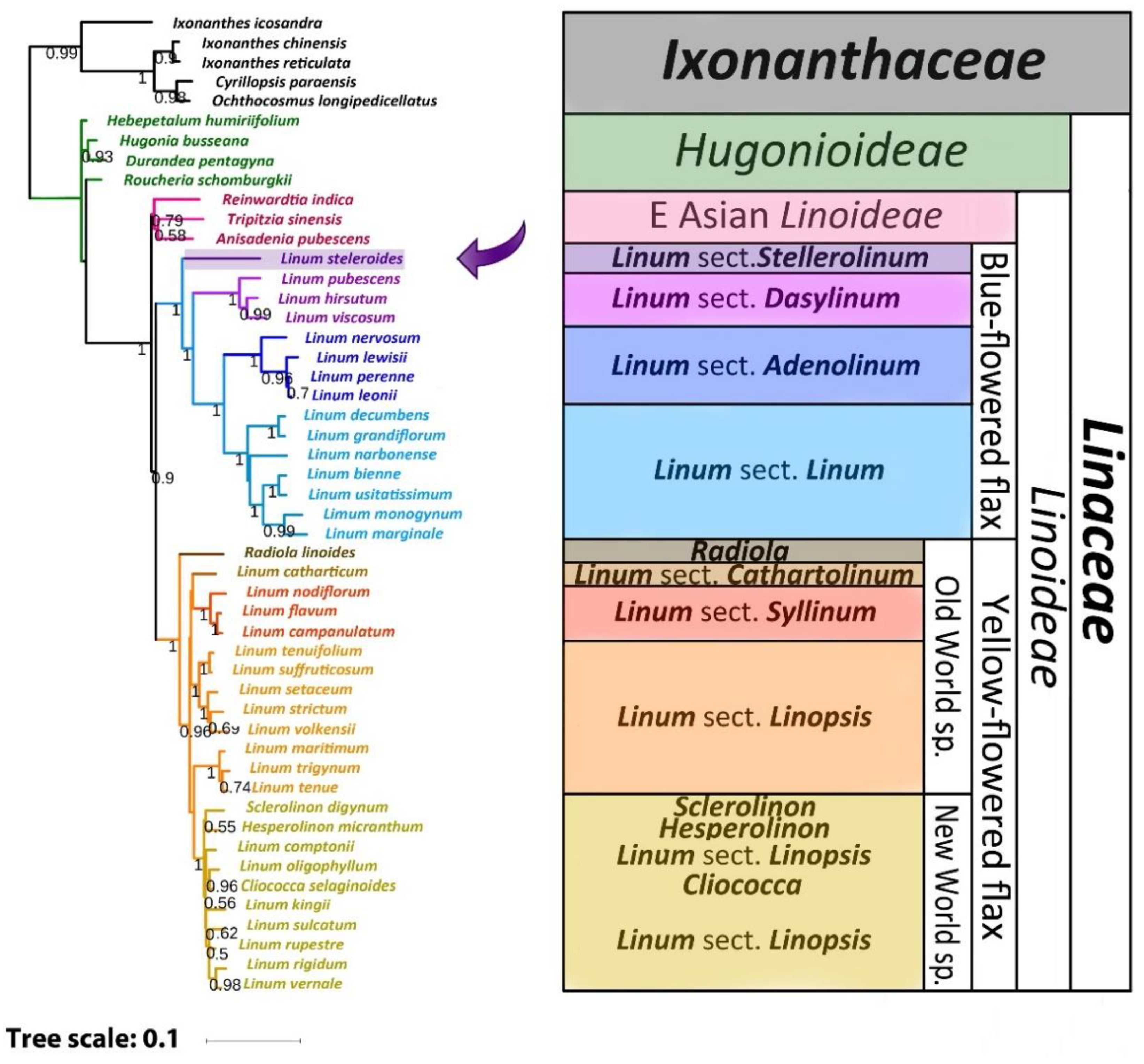
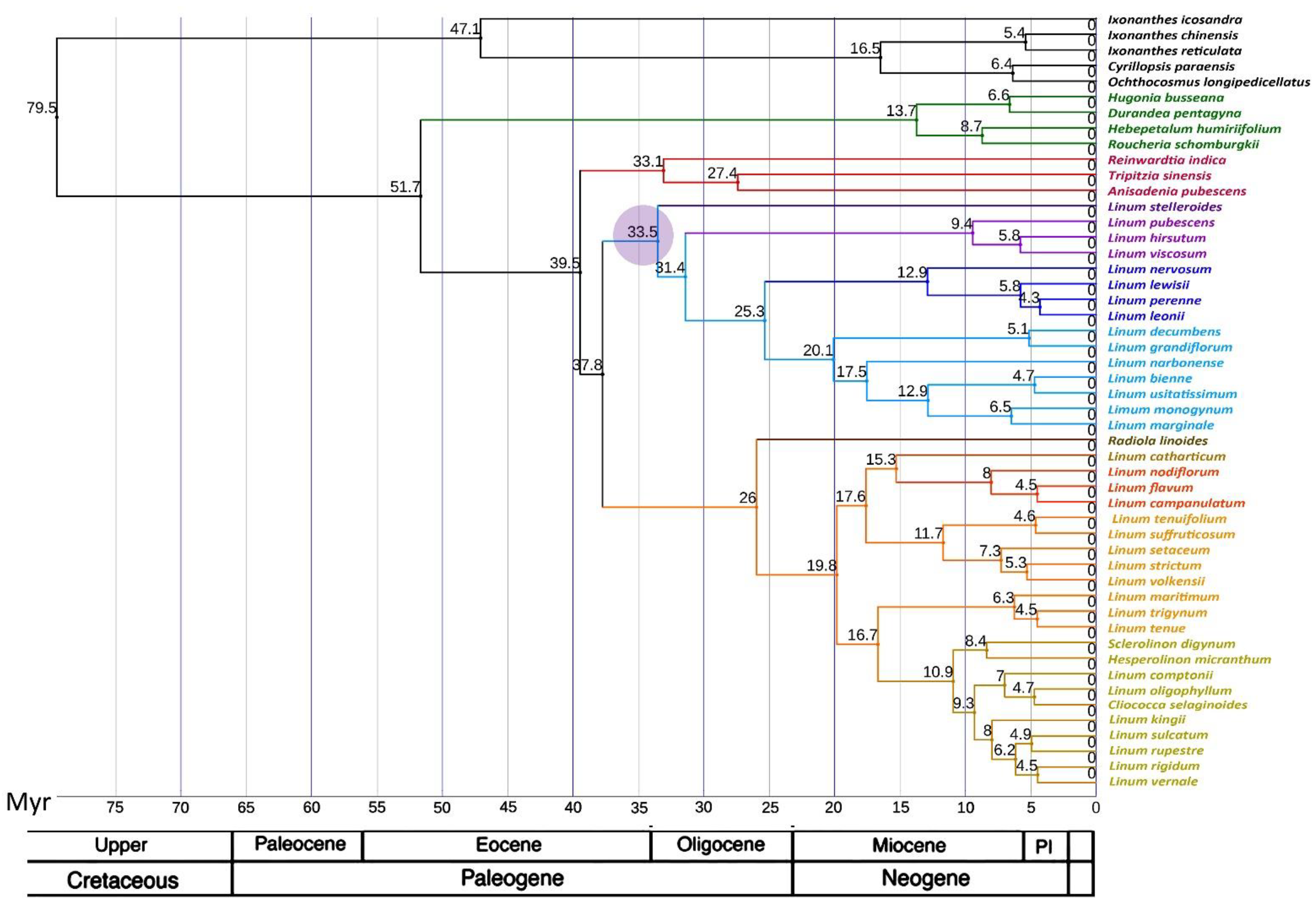
2.3. Phylogenetic Analysis
3. Discussion
3.1. The Reasons for Some Inconsistencies in Previous Phylogenetic Studies
3.2. Comparison of Our Results with Previous Phylogenetic Studies of Flax
4. Materials and Methods
4.1. Data Sources
4.2. Phylogenetic Analysis and Statistical Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogers, C.M.; Xavier, K.S. Pollen morphology as an aid in determining relationships among some widely separated old world species of Linum. Grana 1971, 11, 55–57. [Google Scholar] [CrossRef][Green Version]
- Yuzepchuk, S.V. Genus Linum-Linaceae Dumort. In Flora SSSR (Flora of the Soviet Union); Shishkin, B.K., Bobrov, E.G., Eds.; Izdatel’stvo Akademii Nauk SSSR: Moscow, Russia, 1949; Volume 14, pp. 84–146. [Google Scholar]
- Ockendon, D.J.; Walters, S.M. Linaceae. In Flora Europaea. Rosacea to Umbelliferaceae; Tutin, T.G., Ed.; Cambridge University Press: Cambridge, UK, 1968; Volume 2, pp. 206–211. [Google Scholar]
- Quanru, L.; Zhou, L. Linaceae. In Flora of China. Oxalidaceae through Aceraceae; Wu, Z.Y., Raven, P.H., Eds.; Missouri Botanical Garden Press: St. Louis, MO, USA, 2008; Volume 11, pp. 34–38. [Google Scholar]
- McDill, J.; Repplinger, M.; Simpson, B.; Kadereit, J. The Phylogeny of Linum and Linaceae Subfamily Linoideae, with Implications for Their Systematics, Biogeography, and Evolution of Heterostyly. Syst. Bot. 2009, 34, 386–405. [Google Scholar] [CrossRef]
- Fu, Y.B.; Dong, Y.; Yang, M.H. Multiplexed shotgun sequencing reveals congruent three-genome phylogenetic signals for four botanical sections of the flax genus Linum. Mol. Phylogenetics Evol. 2016, 101, 122–132. [Google Scholar] [CrossRef]
- Fu, Y.-B.; Allaby, R.G. Phylogenetic network of Linum species as revealed by non-coding chloroplast DNA sequences. Genet. Resour. Crop Evol. 2010, 57, 667–677. [Google Scholar] [CrossRef]
- Vromans, J. Molecular Genetic Studies in Flax (Linum Usitatissimum L.). Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2006. [Google Scholar]
- Zhang, J.; Qi, Y.; Wang, L.; Wang, L.; Yan, X.; Dang, Z.; Li, W.; Zhao, W.; Pei, X.; Li, X.; et al. Genomic Comparison and Population Diversity Analysis Provide Insights into the Domestication and Improvement of Flax. iScience 2020, 23, 100967. [Google Scholar] [CrossRef]
- Bolsheva, N.L.; Melnikova, N.V.; Kirov, I.V.; Speranskaya, A.S.; Krinitsina, A.A.; Dmitriev, A.A.; Belenikin, M.S.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; et al. Evolution of blue-flowered species of genus Linum based on high-throughput sequencing of ribosomal RNA genes. BMC Evol. Biol. 2017, 17, 253. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, N.V.; Kudryavtseva, A.V.; Zelenin, A.V.; Lakunina, V.A.; Yurkevich, O.Y.; Speranskaya, A.S.; Dmitriev, A.A.; Krinitsina, A.A.; Belenikin, M.S.; Uroshlev, L.A.; et al. Retrotransposon-based molecular markers for analysis of genetic diversity within the genus Linum. BioMed Res. Int. 2014, 2014, 231589. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.C.; Freyman, W.A.; Guilliams, C.M.; Springer, Y.P.; Baldwin, B.G. Pleistocene radiation of the serpentine-adapted genus Hesperolinon and other divergence times in Linaceae (Malpighiales). Am. J. Bot. 2016, 103, 221–232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Semenova, O.Y.; Samatadze, T.E.; Zelenin, A.V.; Muravenko, O.V. The comparative genome study of the species of Adenolinum and Stellerolinum sections by means of FISH technique. Biol. Membr. 2006, 23, 453–460. [Google Scholar]
- Sokolovskaya, A.P.; Probatova, N.S. Chromosome numbers in the vascular plants from the Primorye territory, Kamchatka, region Amur valley and Sakhalin. Bot. Zhurnal SSSR 1985, 70, 997–999. [Google Scholar]
- Novak, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Cavagnetto, C.; Anadón, P. Preliminary palynological data on floristic and climatic changes during the Middle Eocene-Early Oligocene of the eastern Ebro Basin, northeast Spain. Rev. Palaeobot. Palynol. 1996, 92, 281–305. [Google Scholar] [CrossRef]
- Xi, Z.; Ruhfel, B.R.; Schaefer, H.; Amorim, A.M.; Sugumaran, M.; Wurdack, K.J.; Endress, P.K.; Matthews, M.L.; Stevens, P.F.; Mathews, S.; et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. USA 2012, 109, 17519–17524. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolsheva, N.L.; Melnikova, N.V.; Dvorianinova, E.M.; Mironova, L.N.; Yurkevich, O.Y.; Amosova, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Muravenko, O.V. Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L. Plants 2022, 11, 652. https://doi.org/10.3390/plants11050652
Bolsheva NL, Melnikova NV, Dvorianinova EM, Mironova LN, Yurkevich OY, Amosova AV, Krasnov GS, Dmitriev AA, Muravenko OV. Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L. Plants. 2022; 11(5):652. https://doi.org/10.3390/plants11050652
Chicago/Turabian StyleBolsheva, Nadezhda L., Nataliya V. Melnikova, Ekaterina M. Dvorianinova, Liudmila N. Mironova, Olga Y. Yurkevich, Alexandra V. Amosova, George S. Krasnov, Alexey A. Dmitriev, and Olga V. Muravenko. 2022. "Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L." Plants 11, no. 5: 652. https://doi.org/10.3390/plants11050652
APA StyleBolsheva, N. L., Melnikova, N. V., Dvorianinova, E. M., Mironova, L. N., Yurkevich, O. Y., Amosova, A. V., Krasnov, G. S., Dmitriev, A. A., & Muravenko, O. V. (2022). Clarification of the Position of Linum stelleroides Planch. within the Phylogeny of the Genus Linum L. Plants, 11(5), 652. https://doi.org/10.3390/plants11050652