

Analysis of Genetic Diversity and Resistance to Foliar Pathogens Based on Genotyping-by-Sequencing of a Para Rubber Diversity Panel and Progeny of an Interspecific Cross

 ,
,
Abstract
1. Introduction
2. Results
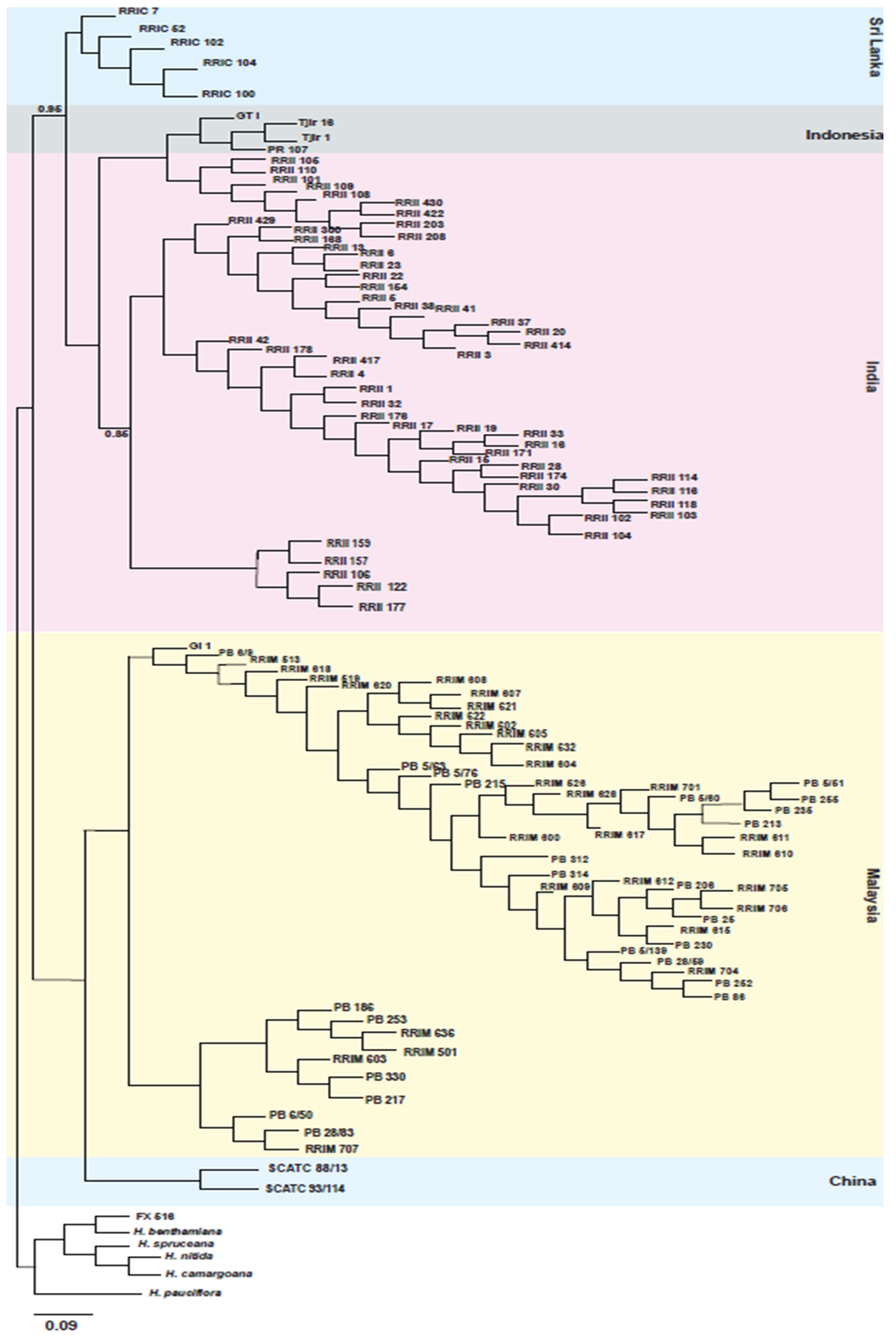
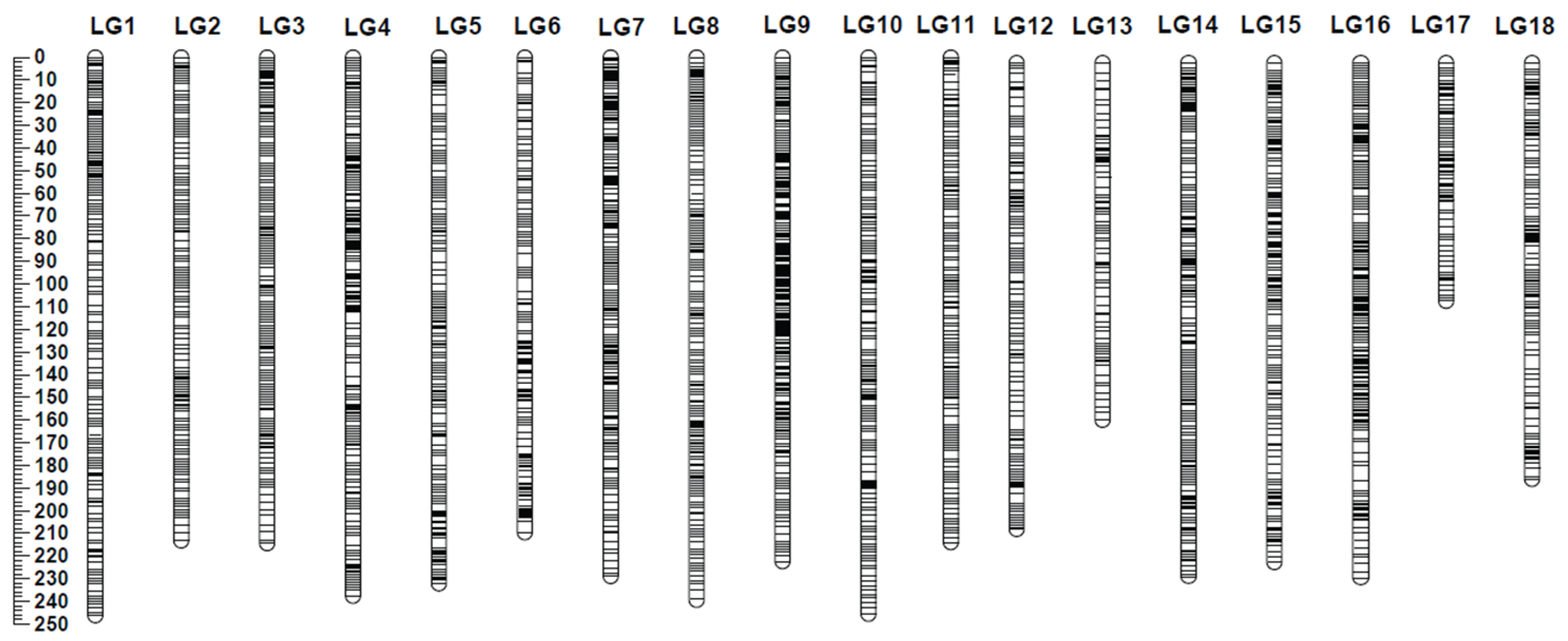
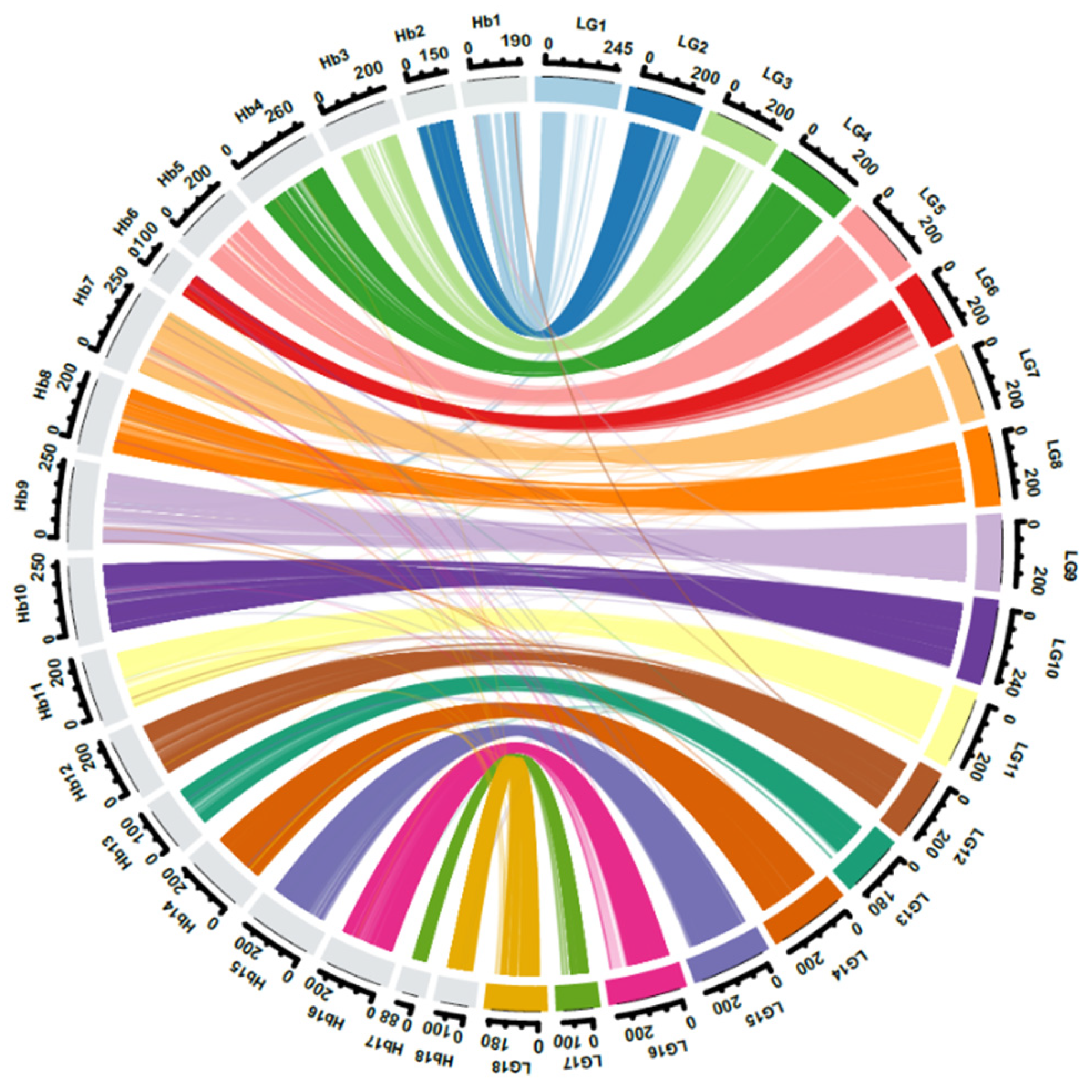
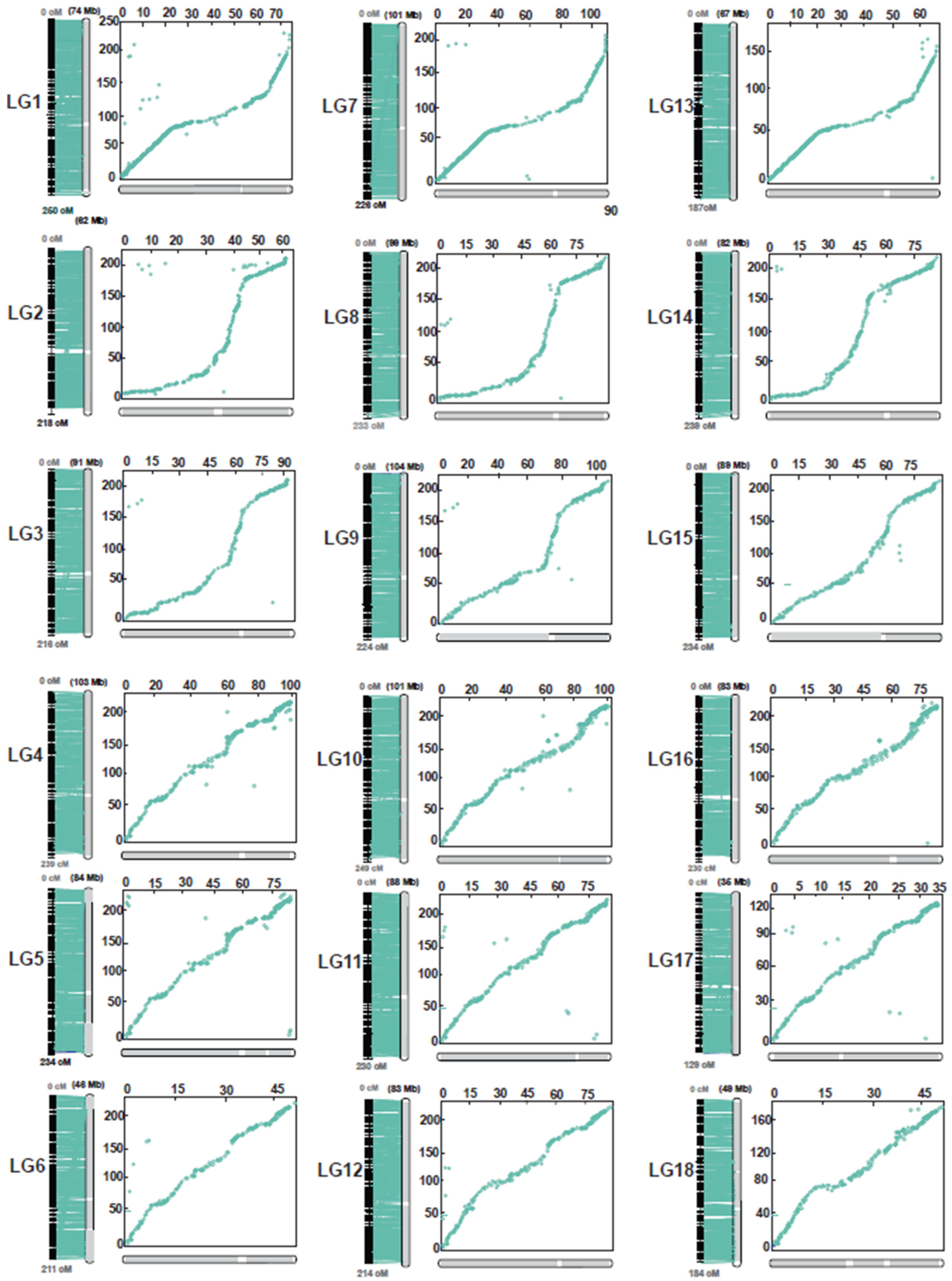
2.1. Genotyping-by-Sequencing, Phylogenetic Analysis, and Linkage Mapping
2.2. Disease Resistance
2.3. Significantly Associated Genes
3. Discussion
4. Materials and Methods
4.1. Diversity Panel and H. brasiliensis (RRII 105) × H. benthamiana (F4542) Mapping Population
4.2. DNA Extraction and DArTSeq Genotyping-By-Sequencing
4.3. Phylogenetic Analysis
4.4. Construction of Integrated Genetic Map Using Lep-Map3 Software
4.5. Genome Scaffold Anchoring and Comparative Mapping between the Integrated Map and the Rubber Reference Genome
4.6. Phenotypic Trait Measurement for Disease Resistance
4.6.1. Screening for Phytophthora Resistance Using Zoospore Suspension
4.6.2. Toxin-Based Screening for Corynespora and Colletotrichum Resistance
4.7. Marker Trait Association Analysis Using Integrated Map
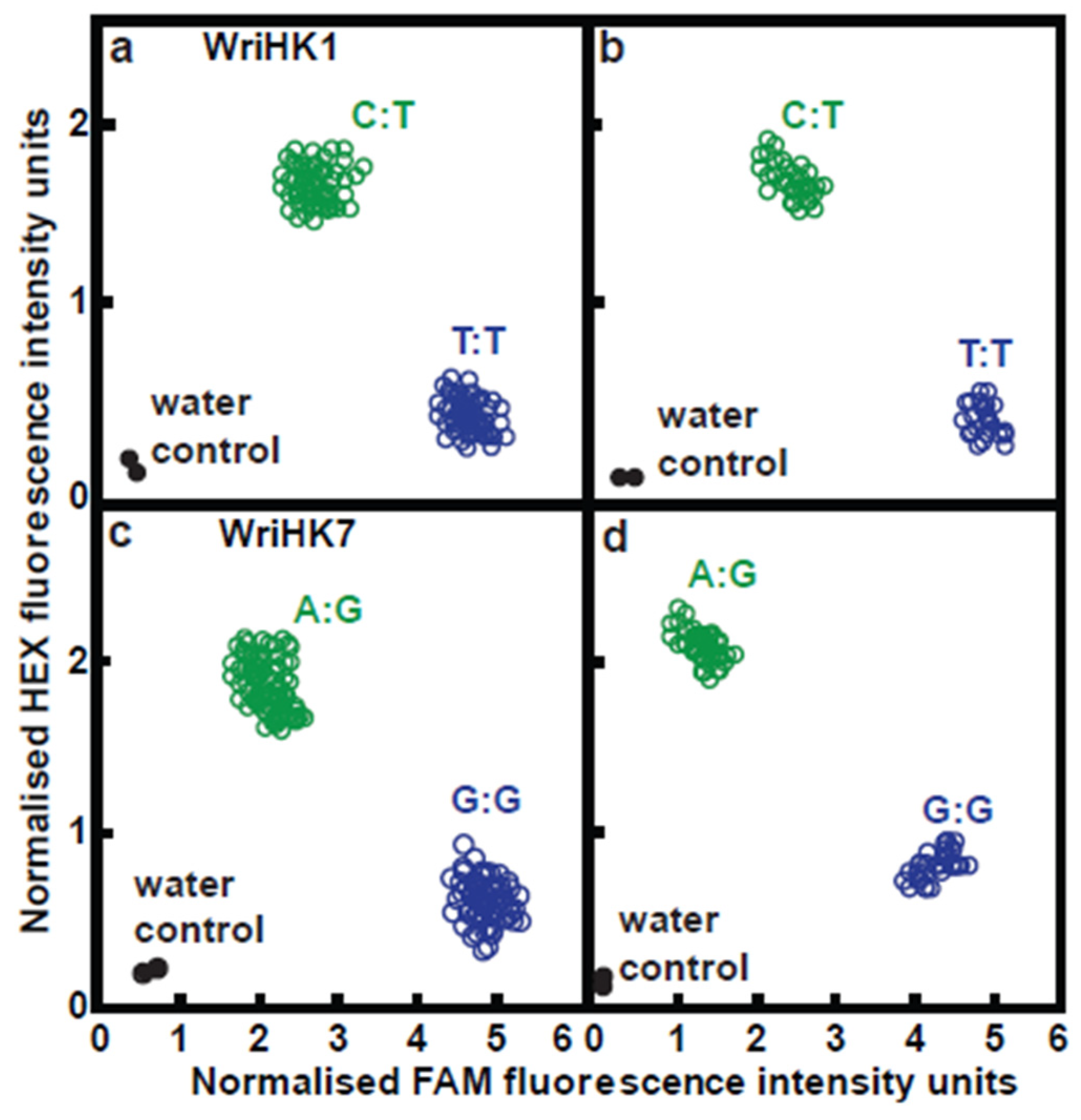
4.8. Development and Use of KASP Assays Where Significant Marker-Trait Association Were Detected
4.9. Candidate QTL Regions and Potential Key Genes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, T.; Priyadarshan, P.M. Genomics of Hevea rubber. In Genomics of Tree Crops; Schnell, R.J., Priyadarshan, P.M., Eds.; Springer: New York, NY, USA, 2012; pp. 261–298. [Google Scholar] [CrossRef]
- Schultes, R.E. Wild Hevea-an untapped source of germplasm. J. Rubber Res. Inst. Sri Lanka 1977, 54, 1–31. [Google Scholar]
- Djikman, M.J. Hevea Thirty Years of Research in the Far East; University of Miami Press: Corel Gables, FL, USA, 1953. [Google Scholar]
- Baulkwill, W.J. The history of natural rubber production. In Rubber; Webster, C.C., Baulkwill, W.J., Eds.; England Longman Scientific and Technical: London, UK, 1989; pp. 1–56. [Google Scholar]
- Simmonds, N.W. Rubber Breeding. In Rubber; Webster, C.C., Baulkwill, W.J., Eds.; England Longman Scientific and Technical: London, UK, 1989; pp. 85–124. [Google Scholar]
- Sagaff, S.S.A.; Ali, S.N.; Mahyudin, M.M.; Wong, M.Y.; Yusop, M.R. Emerging and existing major leaf diseases of Hevea brasiliensis in Malaysia. J. Curr. Opin. Crop Sci. 2022, 3, 34–47. [Google Scholar]
- Mazlan, S.; Jaafar, N.M.; Wahab, A.; Sulaiman, Z.; Rajandas, H.; Zulperi, D. Major diseases of rubber (Hevea brasiliensis) in Malaysia. Pertanika J. Sch. Res. Rev. 2019, 5, 1021. [Google Scholar]
- Ramakrishnan, T.S. Experiments on the control of abnormal leaf fall on Hevea caused by Phytophthora palmivora in South India. In Proceedings of the Natural Rubber Conference, Kuala Lumpur, Malaysia, 26 September–1 October 1960; Rubber Research Institute of Malaya: Kuala Lumpur, Malaysia, 1960; pp. 454–466. [Google Scholar]
- Ogbebor, N.O.; Adekunle, A.T.; Enobakhare, D.A. Inhibition of Colletotrichum gloeosporioides (Penz) Sac. causal organism of rubber (Hevea brasiliensis Muell. Arg.) leaf spot using plant extracts. Afr. J. Biotechnol. 2007, 6, 213–218. [Google Scholar]
- Qu, W.; Li, Z.; Huang, G.; Lin, C.; Ni, W. The current and future potential geographic range of Corynespora Leaf Fall Disease in China. Sens. Lett. 2012, 10, 439–446. [Google Scholar] [CrossRef]
- Lespinasse, D.; Rodier-Goud, M.; Grivet, L.; Leconte, A.; Legnate, H.; Seguin, M. A saturated genetic linkage map of rubber tree (Hevea spp.) based on RFLP, AFLP, microsatellite, and isozyme markers. Theor. Appl. Genet. 2000, 100, 127–138. [Google Scholar] [CrossRef]
- Feng, S.P.; Wei-Guo, L.; Fei, Y.; Jing-Yi, W.; Yao-Ting, W. Construction of genetic linkage map for rubber tree (Hevea brasiliensis) based on SSR markers. Hereditas 2010, 32, 857–863. [Google Scholar]
- Le Guen, V.; Garcia, D.; Doaré, F.; Mattos, C.R.R.; Condina, V.; Couturier, C.; Chambon, A.; Weber, C.; Espéout, S.; Seguin, M. A rubber tree’s durable resistance to Microcyclus ulei is conferred by a qualitative gene and a major quantitative resistance factor. Tree Genet. Genom. 2011, 7, 877–889. [Google Scholar] [CrossRef]
- Triwitayakorn, K.; Chatkulkawin, P.; Kanjanawattanawong, S.; Sraphet, S.; Yoocha, T.; Sangsrakru, D. Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res. 2011, 18, 471–482. [Google Scholar] [CrossRef]
- Souza, L.M.; Mantello, C.C.; Suzuki, F.; Gazaffi, R.; Garcia, D.; Le Guen, V.; Garcia, A.A.F.; Souza, A.P. Development of a genetic linkage map of rubber tree (Hevea braziliensis) based on microsatellite markers. BMC Proc. 2011, 5, 39. [Google Scholar] [CrossRef]
- Souza, L.M.; Gazaffi, R.; Mantello, C.C.; Silva, C.C.; Garcia, D.; Le Guen, V. QTL mapping of growth-related traits in a full-sib family of rubber tree (Hevea brasiliensis) evaluated in a sub-tropical climate. PLoS ONE 2013, 8, e61238. [Google Scholar] [CrossRef] [PubMed]
- Pootakham, W.; Ruang-Areerate, P.; Jomchai, N.; Sonthirod, C.; Sangsrakru, D.; Yoocha, T.; Theerawattanasuk, K.; Nirapathpongporn, K.; Romruensukharom, P.; Tragoonrung, S.; et al. Construction of a high-density integrated genetic linkage map of rubber tree (Hevea brasiliensis) using genotyping-by-sequencing (GBS). Front. Plant Sci. 2015, 6, 367. [Google Scholar] [CrossRef]
- Conson, A.R.O.; Taniguti, C.H.; Amadeu, R.R.; Andreotti, I.A.A.; de Souza, L.M.; dos Santos, L.H.B.; Rosa, J.R.B.F.; Mantello, C.C.; da Silva, C.C.; Junior, E.J.S.; et al. High resolution genetic map and QTL analysis of growth-related traits of Hevea brasiliensis cultivated under suboptimal temperature and humidity conditions. Front. Plant Sci. 2018, 9, 1255. [Google Scholar] [CrossRef] [PubMed]
- Francisco, F.R.; Aono, A.H.; da Silva, C.C.; Gonçalves, P.S.; Scaloppi Junior, E.J.; Le Guen, V.; Fritsche-Neto, R.; Souza, L.M.; Souza, A.P. Unravelling rubber tree growth by integrating GWAS and biological network-based approaches. Front. Plant Sci. 2021, 12, 768589. [Google Scholar] [CrossRef] [PubMed]
- Rastas, P. Lep-MAP3: Robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 2017, 33, 3726–3732. [Google Scholar] [CrossRef]
- Liu, J.; Shi, C.; Li, W.; Zhang, Q.; Zhang, Y. The chromosome-based rubber tree genome provides new insights into spurge genome evolution and rubber biosynthesis. Mol. Plant 2020, 13, 336–350. [Google Scholar] [CrossRef]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Ruang-Areerate, P.; Yoocha, T.; Sangsrakru, D. De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci. Rep. 2017, 7, 41457. [Google Scholar] [CrossRef]
- He, C.; Holme, J.; Anthony, J. SNP genotyping: The KASP assay. In Crop Breeding; Humana Press: New York, NY, USA, 2014; pp. 75–86. [Google Scholar]
- Perseguini, J.M.K.C.; Romão, L.R.C.; Briñez, B.; Junior, E.J.S.; Gonçalves, P.S.; Benchimol, L.L. Genetic diversity of cultivated accessions and wild species of rubber tree using EST-SSR markers. Pesq. Agropec. Bras. Brasília 2012, 47, 1087–1094. [Google Scholar] [CrossRef]
- Narayanan, C.; Mydin, K.K. Breeding for disease resistance in Hevea spp.-Status, potential threats, and possible strategies. In Proceedings of the 4th International Workshop on Genetics of Host-Parasite Interactions in Forestry: Disease and Insect Resistance in Forest Trees, General Technical report PSW-GTR-240, Valley River Inn, Eugene, OR, USA, 31 July–5 August 2011; pp. 240–251. [Google Scholar]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef]
- Goonetilleke, S.N.; March, T.J.; Wirthensohn, M.G.; Arús, P.; Walker, A.R.; Mather, D.E. Genotyping-by-sequencing in almond: SNP discovery, linkage mapping and marker design. Genes Genomes Genet. 2018, 8, 161–172. [Google Scholar] [CrossRef]
- Wastie, R.L. Nursery screening of Hevea for resistance to Gleosporium leaf disease. J. Rubber Res. 1973, 23, 339. [Google Scholar]
- Lim, T.M. A rapid laboratory method of assessing susceptibility of Hevea clones to Oidium Hevea. Exp. Agric. 1973, 9, 275. [Google Scholar] [CrossRef]
- Tan, A.M.; Loo, T.P.; Vadivel, G.; Ros, M.; Bach, K.; Yoon, K.F. Survey of major leaf diseases of rubber in Peninsular Malaysia. Plant. Bull. Rubber Res. Inst. Malays. 1992, 2, 51. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Roy, C.B.; Ravindran, M.; Saha, T. Efficient screening of AFLP primer combinations for evaluating genetic diversity among cultivated rubber (Hevea brasiliensis) clones. Nat. Rubber Res. 2012, 25, 21–30. [Google Scholar]
- Rambaut, A. FigTree v1.4.2. A Graphical Viewer of Phylogenetic Trees. 2018. Available online: https://tree.bio.ed.ac.uk/software/figtree/ (accessed on 22 November 2022).
- Milne, I.; Shaw, P.; Stephen, G.; Bayer, M.; Cardle, L.; Thomas, W.T.B.; Flavell, A.J.; Marshall, D. Flapjack-graphical genotype visualization. Bioinformatics 2010, 26, 3133–3134. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Feng, L.Y.; Liu, J.; Gao, C.W.; Wu, H.B.; Gao, L.Z. Higher genomic variation in wild than cultivated rubber trees, Hevea brasiliensis, revealed by comparative analyses of chloroplast genomes. Front. Ecol. Evol. 2020, 8, 237–247. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Rajalakshmy, V.K.; Joseph, A. Production of sporangia by Phytophthora meadii. Indian Phytopathol. 1986, 39, 470–471. [Google Scholar]
- Joseph, A.; Mathew, F.; Mercy, M.A. Evaluation of wild Hevea germplasm against Phytophthora meadii causing abnormal leaf fall disease of rubber in India. In Proceedings of the International Rubber Workshop on Hevea Diseases in Africa, Benin City, Nigeria, 5–7 June 2012; pp. 55–65. [Google Scholar]
- Borjesson, T.; Stollman, U.; Schnurer, T. Volatile metabolites and other indicators of Penicillium aurantiogriseum growth on different substrates. Appl. Environ. Microbiol. 1990, 56, 3705–3710. [Google Scholar] [CrossRef] [PubMed]
- Breton, F.; Sanier, C.; D’Auzac, J. Role of cassiicolin, a host-selective toxin, in pathogenicity of Corynespora cassiicola, causal agent of a leaf fall disease of Hevea. J. Rubber Res. 2000, 3, 115–128. [Google Scholar]
- Hallé, F.; Martin, R. Etude de la croissance rythmique chez l’hévéa (Hevea brasiliensis) Müll. Arg., (Euphorbiacées, crotonoïdées). Adansonia 1968, 8, 475–503. [Google Scholar]
- Fernando, T.H.P.S.; Jayasinghe, C.K.; Wijesundera, R.L.C.; Silva, W.P.K.; Nishantha, E.A.D.N. Evaluation of screening methods against Corynespora leaf fall disease of rubber (Hevea brasiliensis). J. Plant Dis. Prot. 2010, 117, 24–29. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Schulze, T.G.; McMahon, F.J. Genetic association mapping at the crossroads: Which test and why? Overview and practical guidelines. Am. J. Med. Genet. 2002, 114, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Colletotrichum acutatum Resistance Classification | Corynespora cassiicola Resistance Classification | Total | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 1 | 2 | 2 | 2 | 1 | 2 | 9 (11.3%) |
| 2 | 4 | 8 | 5 | 6 | 3 | 26 (32.9%) |
| 3 | 1 | 9 | 4 | 6 | 5 | 25 (31.6%) |
| 4 | 1 | 5 | 3 | 4 | 1 | 14 (17.8%) |
| 5 | 0 | 1 | 2 | 1 | 1 | 5 (6.4%) |
| Total | 8 (10.1%) | 25 (31.6%) | 16 (20.2%) | 18 (22.8%) | 12 (15.2%) | 79 (100%) |
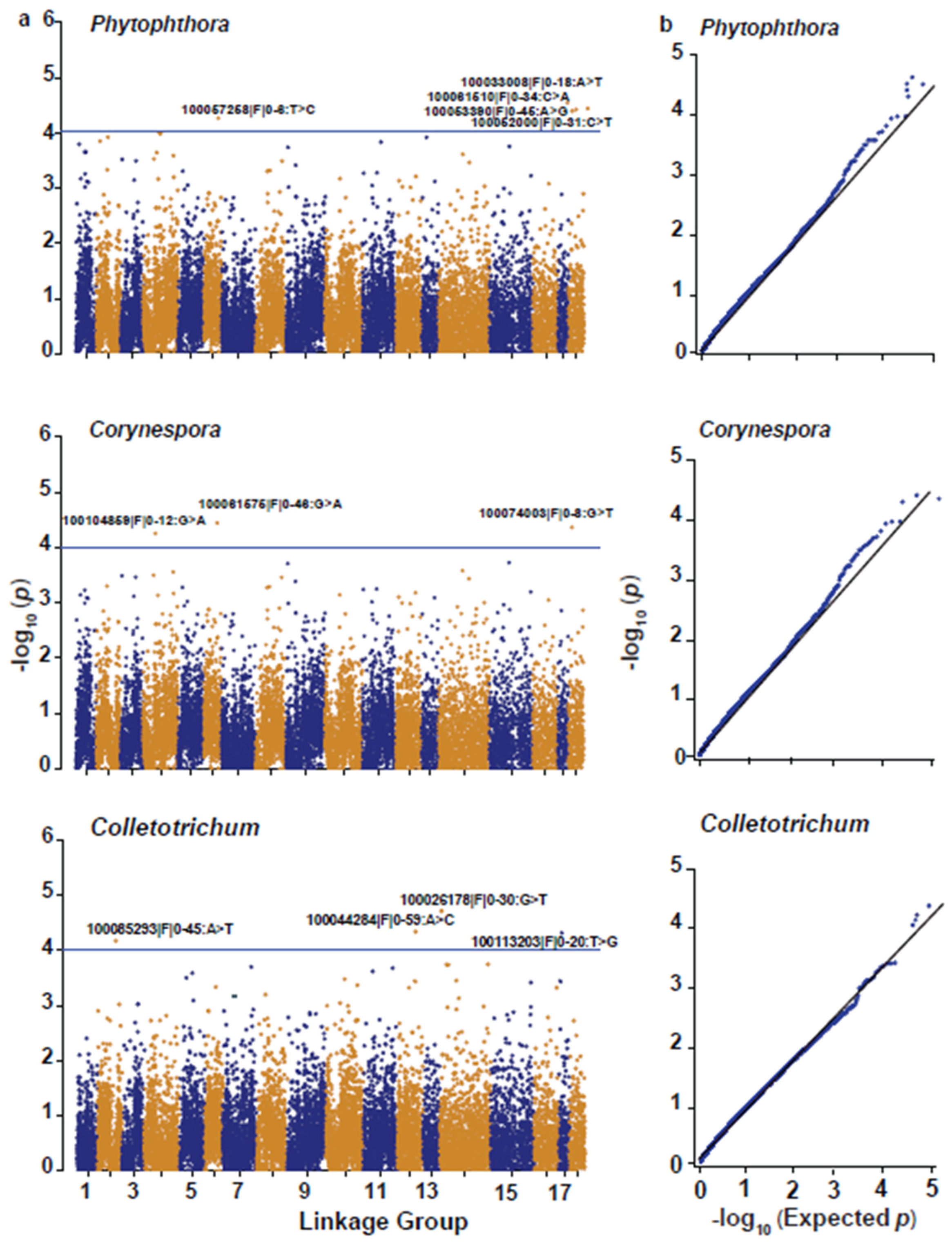
| Pathogen Associated with the Disease Trait | Linkage Group | Marker | Reference Genome Sequence | Physical Position (bp) | −Log10 (p) | R2 (%) (Variance Explained) | Allele Effect | |
|---|---|---|---|---|---|---|---|---|
| Start | End | |||||||
| Phytophthora | LG6 | 100057258|F|0–6:T > C | KB619684.1_scaffold 218036 | 28,834 | 28,903 | 4.28 | 14 | 1.10 |
| LG18 | 100033008|F|0–18:A > T | GT1 Reference genome (CM021243.1) | 18,128,717 | 18,128,649 | 4.52 | 18 | 3.21 | |
| 100061510|F|0–34:C > A | GT1 Reference genome (CM021243.1) | 12,918,745 | 12,918,685 | 4.25 | 14 | 2.57 | ||
| 100053390|F|0–45:A > G | GT1 Reference genome (CM021243.1) | 15,754,360 | 15,754,413 | 4.25 | 14 | 2.57 | ||
| 100052000|F|0–31:C > T | GT1 Reference genome (CM021243.1) | 15,953,978 | 15,953,939 | 4.21 | 14 | 2.32 | ||
| Corynespora | LG4 | 100104859|F|0–12:G > A | GT1 Reference genome (CM021229.1) | 34,907,019 | 34,906,974 | 4.21 | 14 | 2.30 |
| LG6 | 100061575|F|0–46:G > A | GT1 Reference genome (CM021231.1) | 37,466,880 | 37,466,948 | 4.35 | 16 | 2.43 | |
| LG18 | 100074003|F|0–8:G > T | GT1 Reference genome (CM021243.1) | 44,256,460 | 44,256,528 | 4.35 | 16 | 2.41 | |
| Colletotrichum | LG2 | 100085293|F|0–45:A > T | AJJZ010325919.1_contig 401252 | 1511 | 1580 | 4.13 | 11 | 1.25 |
| LG12 | 100044284|F|0–59:A > C | GT1 Reference genome (CM021237.1) | 73,830,891 | 73,830,823 | 4.26 | 14 | 1.56 | |
| LG14 | 100026178|F|0–30:G > T | GT1 Reference genome (CM021239.1) | 6,682,648 | 6,682,714 | 4.48 | 16 | 0.98 | |
| LG17 | 100113203|F|0–20:T > G | GT1 Reference genome (CM021242.1) | 2,385,929 | 2,385,861 | 4.21 | 16 | 0.97 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, C.B.; Goonetilleke, S.N.; Joseph, L.; Krishnan, A.; Saha, T.; Kilian, A.; Mather, D.E. Analysis of Genetic Diversity and Resistance to Foliar Pathogens Based on Genotyping-by-Sequencing of a Para Rubber Diversity Panel and Progeny of an Interspecific Cross. Plants 2022, 11, 3418. https://doi.org/10.3390/plants11243418
Roy CB, Goonetilleke SN, Joseph L, Krishnan A, Saha T, Kilian A, Mather DE. Analysis of Genetic Diversity and Resistance to Foliar Pathogens Based on Genotyping-by-Sequencing of a Para Rubber Diversity Panel and Progeny of an Interspecific Cross. Plants. 2022; 11(24):3418. https://doi.org/10.3390/plants11243418
Chicago/Turabian StyleRoy, C. Bindu, Shashi N. Goonetilleke, Limiya Joseph, Anu Krishnan, Thakurdas Saha, Andrzej Kilian, and Diane E. Mather. 2022. "Analysis of Genetic Diversity and Resistance to Foliar Pathogens Based on Genotyping-by-Sequencing of a Para Rubber Diversity Panel and Progeny of an Interspecific Cross" Plants 11, no. 24: 3418. https://doi.org/10.3390/plants11243418
APA StyleRoy, C. B., Goonetilleke, S. N., Joseph, L., Krishnan, A., Saha, T., Kilian, A., & Mather, D. E. (2022). Analysis of Genetic Diversity and Resistance to Foliar Pathogens Based on Genotyping-by-Sequencing of a Para Rubber Diversity Panel and Progeny of an Interspecific Cross. Plants, 11(24), 3418. https://doi.org/10.3390/plants11243418