
Multiomic Data Integration in the Analysis of Drought-Responsive Mechanisms in Quercus ilex Seedlings

 , , , , and
, , , , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
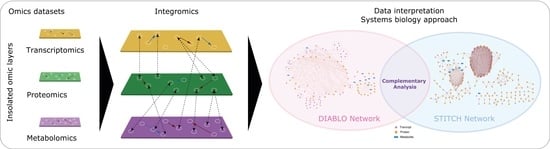
2.1. Comparison of Principal Component Analysis (PCA) and DIABLO Multiomic Data Analysis
2.2. DIABLO and STITCH Interaction Networks and Their Biological Interpretation
3. Materials and Methods
3.1. Plant Material and Experiment Description
3.2. Non-Targeted Transcriptomic Analysis
3.3. Non-Targeted Proteomic Analysis
3.4. Non-Targeted Metabolomics Analysis
3.5. Data Preprocessing and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patón, D.; García-Herrera, R.; Cuenca, J.; Galavis, M.; Roig, F. Influence of Climate on Radial Growth of Holm Oaks (Quercus ilex Subsp. Ballota Desf) from SW Spain. Geochronometria 2010, 34, 49–56. [Google Scholar]
- Haavik, L.J.; Billings, S.A.; Guldin, J.M.; Stephen, F.M. Emergent insects, pathogens and drought shape changing patterns in oak decline in North America and Europe. For. Ecol. Manag. 2015, 354, 190–205. [Google Scholar]
- Natalini, F.; Alejano, R.; Vázquez-Piqué, J.; Cañellas, I.; Gea-Izquierdo, G. The role of climate change in the widespread mortality of holm oak in open woodlands of Southwestern Spain. Dendrochronologia 2016, 38, 51–60. [Google Scholar] [CrossRef]
- Villar-Salvador, P.; Planelles, R.; Oliet, J.; Peñuelas-Rubira, J.L.; Jacobs, D.F.; González, M. Drought tolerance and transplanting performance of holm oak (Quercus ilex) seedlings after drought hardening in the nursery. Tree Physiol. 2004, 24, 1147–1155. [Google Scholar]
- Keenan, T.F.; Serra, J.M.; Lloret, F.; Ninyerola, M.; Sabaté, S. Predicting the future of forests in the Mediterranean under climate change, with niche- and process-based models: CO2 matters! Glob. Chang. Biol. 2011, 17, 565–579. [Google Scholar]
- Ogaya, R.; Liu, D.; Barbeta, A.; Peñuelas, J. Stem Mortality and Forest Dieback in a 20-Years Experimental Drought in a Mediterranean Holm Oak Forest. Front. For. Glob. Chang. 2020, 2, 89. [Google Scholar] [CrossRef]
- San-Eufrasio, B.; Sánchez-Lucas, R.; López-Hidalgo, C.; Guerrero-Sánchez, V.M.; Castillejo, M.; Maldonado-Alconada, A.M.; Jorrín-Novo, J.V.; Rey, M.-D. Responses and differences in tolerance to water shortage under climatic dryness conditions in seedlings from Quercus spp. and Andalusian Q. ilex populations. Forests 2020, 11, 707. [Google Scholar]
- Tienda-Parrilla, M.; López-Hidalgo, C.; Guerrero-Sanchez, V.M.; Infantes-González, Á.; Valderrama-Fernández, R.; Castillejo, M.-Á.; Jorrín-Novo, J.V.; Rey, M.-D. Untargeted MS-Based Metabolomics Analysis of the Responses to Drought Stress in Quercus ilex L. Leaf Seedlings and the Identification of Putative Compounds Related to Tolerance. Forests 2022, 13, 551. [Google Scholar] [CrossRef]
- Rey, M.-D.; Castillejo, M.Á.; Sánchez-Lucas, R.; Guerrero-Sanchez, V.M.; López-Hidalgo, C.; Romero-Rodríguez, C.; Valero-Galván, J.; Sghaier-Hammami, B.; Simova-Stoilova, L.; Echevarría-Zomeño, S.; et al. Proteomics, holm oak (Quercus ilex L.) and other recalcitrant and orphan forest tree species: How do they see each other? Int. J. Mol. Sci. 2019, 20, 692. [Google Scholar] [CrossRef]
- Maldonado-Alconada, A.M.; Castillejo, M.Á.; Rey, M.-D.; Labella-Ortega, M.; Tienda-Parrilla, M.; Hernández-Lao, T.; Honrubia-Gómez, I.; Ramírez-García, J.; Guerrero-Sanchez, V.M.; López-Hidalgo, C.; et al. Multiomics Molecular Research into the Recalcitrant and Orphan Quercus ilex Tree Species: Why, What for, and How. Int. J. Mol. Sci. 2022, 23, 9980. [Google Scholar] [CrossRef]
- Rey, M.; Labella-Ortega, M.; Guerrero-Sánchez, V.; Carleial, R.; Castillejo, M.; Rodríguez-Franco, A.; Buggs, R.; Ruggieri, V.; Jorrín-Novo, J. A first draft genome of Holm oak (Quercus ilex L.), the most representative species of the Mediterranean forest and the Spanish agrosilvopastoral ecosystem “dehesa”. bioRxiv 2022. bioRxiv: 511480. [Google Scholar]
- Escandón, M.; Castillejo, M.Á.; Jorrín-Novo, J.V.; Rey, M.-D. Molecular research on stress responses in Quercus spp.: From classical biochemistry to systems biology through Omics analysis. Forests 2021, 12, 364. [Google Scholar] [CrossRef]
- Guerrero-Sánchez, V.M.; Maldonado-Alconada, A.M.; Amil-Ruiz, F.; Verardi, A.; Jorrín-Novo, J.V.; Rey, M.-D. Ion Torrent and lllumina, two complementary RNA-seq platforms for constructing the Holm oak (Quercus ilex) transcriptome. PLoS ONE 2019, 14, e0210356. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Sánchez, V.M.; Castillejo, M.Á.; López-Hidalgo, C.; Alconada, A.M.M.; Jorrín-Novo, J.V.; Rey, M.-D. Changes in the transcript and protein profiles of Quercus ilex seedlings in response to drought stress. J. Proteomics 2021, 243, 104263. [Google Scholar] [CrossRef]
- López-Hidalgo, C.; Trigueros, M.; Menéndez, M.; Jorrin-Novo, J.V. Phytochemical composition and variability in Quercus ilex acorn morphotypes as determined by NIRS and MS-based approaches. Food Chem. 2021, 338, 127803. [Google Scholar] [CrossRef]
- Karahalil, B. Overview of Systems Biology and Omics Technologies. Curr. Med. Chem. 2016, 23, 4221–4230. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.-P.; Bodein, A.; Périn, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021, 19, 3735–3746. [Google Scholar] [CrossRef]
- Fabres, P.J.; Collins, C.; Cavagnaro, T.R.; Rodríguez López, C.M. A Concise Review on Multi-Omics Data Integration for Terroir Analysis in Vitis vinifera. Front. Plant Sci. 2017, 8, 1065. [Google Scholar] [CrossRef]
- Yan, J.; Risacher, S.L.; Shen, L.; Saykin, A.J. Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Brief. Bioinform. 2018, 19, 1370–1381. [Google Scholar] [CrossRef]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef] [PubMed]
- San-Eufrasio, B.; Bigatton, E.; Guerrero-Sanchez, V.; Chaturvedi, P.; Jorrin-Novo, J.; Rey, M.-D.; Castillejo, M. Proteomics data analysis for the identification of proteins and derived proteotypic peptides of potential use as putative drought tolerance markers for Quercus ilex. Int. J. Mol. Sci. 2021, 22, 3191. [Google Scholar] [PubMed]
- San-Eufrasio, B.; Castillejo, M.Á.; Labella-Ortega, M.; Ruiz-Gómez, F.J.; Navarro-Cerrillo, R.M.; Tienda-Parrilla, M.; Jorrín-Novo, J.V.; Rey, M.-D. Effect and Response of Quercus ilex subsp. ballota [Desf.] Samp. Seedlings From Three Contrasting Andalusian Populations to Individual and Combined Phytophthora cinnamomi and Drought Stresses. Front. Plant Sci. 2021, 12, 722802. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Sánchez, V.M.; Maldonado-Alconada, A.M.; Amil-Ruiz, F.; Jorrin-Novo, J. V Holm oak (Quercus ilex) transcriptome. De novo sequencing and assembly analysis. Front. Mol. Biosci. 2017, 4, 70. [Google Scholar] [CrossRef]
- López-Hidalgo, C.; Guerrero-Sánchez, V.M.; Gómez-Gálvez, I.; Sánchez-Lucas, R.; Castillejo-Sánchez, M.A.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrín-Novo, J. V A multi-omics analysis pipeline for the metabolic pathway reconstruction in the orphan species Quercus ilex. Front. Plant Sci. 2018, 9, 935. [Google Scholar] [CrossRef]
- Echevarría-Zomeño, S.; Ariza, D.; Jorge, I.; Lenz, C.; del Campo, A.D.; Jorrín, J.; Navarro, R.M. Changes in the protein profile of Quercus ilex leaves in response to drought stress and recovery. J. Plant Physiol. 2009, 166, 233–245. [Google Scholar] [CrossRef]
- Valero-Galván, J.; González-Fernández, R.; Navarro-Cerrillo, R.; Gil-Pelegrin, E.; Jorrín-Novo, J. Physiological and proteomic analyses of drought stress response in Holm oak provenances. J. Proteome Res. 2013, 12, 5110–5123. [Google Scholar]
- Simova-Stoilova, L.P.; Romero-Rodríguez, M.C.; Sánchez-Lucas, R.; Navarro-Cerrillo, R.M.; Medina-Aunon, J.A.; Jorrín-Novo, J.V. 2-DE proteomics analysis of drought treated seedlings of Quercus ilex supports a root active strategy for metabolic adaptation in response to water shortage. Front. Plant Sci. 2015, 6, 627. [Google Scholar]
- Simova-Stoilova, L.P.; López-Hidalgo, C.; Sanchez-Lucas, R.; Valero-Galvan, J.; Romero-Rodríguez, C.; Jorrin-Novo, J.V. Holm oak proteomic response to water limitation at seedling establishment stage reveals specific changes in different plant parts as well as interaction between roots and cotyledons. Plant Sci. 2018, 276, 1–13. [Google Scholar] [CrossRef]
- Vaseva, I.; Sabotič, J.; Sustar-vozlic, J.; Meglic, V.; Kidrič, M.; Demirevska, K.; Simova, L. The response of plants to drought stress: The role of dehydrins, chaperones, proteases and protease inhibitors in maintaining cellular protein function. In Droughts: New Research; Neves, D.F., Sanz, J.D., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; pp. 1–45. ISBN 978-1-62100-769-2. [Google Scholar]
- Heinemann, B.; Künzler, P.; Eubel, H.; Braun, H.-P.; Hildebrandt, T.M. Estimating the number of protein molecules in a plant cell: Protein and amino acid homeostasis during drought. Plant Physiol. 2021, 185, 385–404. [Google Scholar]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant. Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Close, T.J. Expression of dehydrins under heat stress and their relationship with water relations of sugarcane leaves. Biol. Plant. 2007, 51, 104–109. [Google Scholar]
- Dvořák, P.; Krasylenko, Y.; Zeiner, A.; Šamaj, J.; Takáč, T. Signaling Toward Reactive Oxygen Species-Scavenging Enzymes in Plants. Front. Plant Sci. 2020, 11, 618835. [Google Scholar]
- Kavi Kishor, P.B.; Sreenivasulu, N. Is proline accumulation per se correlated with stress tolerance or is proline homeostasis a more critical issue? Plant. Cell Environ. 2014, 37, 300–311. [Google Scholar] [CrossRef]
- Almeida, T.; Pinto, G.; Correia, B.; Gonçalves, S.; Meijón, M.; Escandón, M. In-depth analysis of the Quercus suber metabolome under drought stress and recovery reveals potential key metabolic players. Plant Sci. 2020, 299, 110606. [Google Scholar] [CrossRef]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res. 2008, 36, D684–D688. [Google Scholar] [CrossRef]
- Izhaki, I. Emodin—A secondary metabolite with multiple ecological functions in higher plants. New Phytol. 2002, 155, 205–217. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, B.; Xie, J.; Xu, P.; Zhang, Y.; Ming, J. Effect of enrofloxacin and emodin on heat-shock proteins’ expression in hepatic cells of grass carp (Ctenopharyngodon idellus). Aquac. Int. 2014, 22, 1067–1077. [Google Scholar] [CrossRef]
- Rajendrakumar, C.S.V.; Suryanarayana, T.; Reddy, A.R. DNA helix destabilization by proline and betaine: Possible role in the salinity tolerance process. FEBS Lett. 1997, 410, 201–205. [Google Scholar]
- Takano, K.; Higashi, R.; Okada, J.; Mukaiyama, A.; Tadokoro, T.; Koga, Y.; Kanaya, S. Proline effect on the thermostability and slow unfolding of a hyperthermophilic protein. J. Biochem. 2009, 145, 79–85. [Google Scholar] [PubMed]
- Zelisko, A.; García-Lorenzo, M.; Jackowski, G.; Jansson, S.; Funk, C. AtFtsH6 is involved in the degradation of the light-harvesting complex II during high-light acclimation and senescence. Proc. Natl. Acad. Sci. USA 2005, 102, 13699–13704. [Google Scholar] [CrossRef] [PubMed]
- Adam, Z.; Rudella, A.; van Wijk, K.J. Recent advances in the study of Clp, FtsH and other proteases located in chloroplasts. Curr. Opin. Plant Biol. 2006, 9, 234–240. [Google Scholar] [PubMed]
- Shiraku, M.L.; Magwanga, R.O.; Cai, X.; Kirungu, J.N.; Xu, Y.; Mehari, T.G.; Hou, Y.; Wang, Y.; Wang, K.; Peng, R.; et al. Knockdown of 60S ribosomal protein L14-2 reveals their potential regulatory roles to enhance drought and salt tolerance in cotton. J. Cott. Res. 2021, 4, 27. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef]
- Manna, M.; Thakur, T.; Chirom, O.; Mandlik, R.; Deshmukh, R.; Salvi, P. Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant. 2021, 172, 847–868. [Google Scholar] [CrossRef]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef]
- Kiełbowicz-Matuk, A.; Czarnecka, J.; Banachowicz, E.; Rey, P.; Rorat, T. Solanum tuberosum ZPR1 encodes a light-regulated nuclear DNA-binding protein adjusting the circadian expression of StBBX24 to light cycle. Plant. Cell Environ. 2017, 40, 424–440. [Google Scholar] [CrossRef]
- Bundock, P.; Hooykaas, P. An Arabidopsis hAT-like transposase is essential for plant development. Nature 2005, 436, 282–284. [Google Scholar] [CrossRef]
- Ikeda, M.; Fujiwara, S.; Mitsuda, N.; Ohme-Takagi, M. A triantagonistic basic helix-loop-helix system regulates cell elongation in Arabidopsis. Plant Cell 2012, 24, 4483–4497. [Google Scholar]
- Sakuma, Y.; Maruyama, K.; Qin, F.; Osakabe, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18822–18827. [Google Scholar]
- Jia, J.; Zhou, J.; Shi, W.; Cao, X.; Luo, J.; Polle, A.; Luo, Z. Comparative transcriptomic analysis reveals the roles of responsive genes in poplars exposed to high temperature and drought. Nature 2017, 7, 43215. [Google Scholar]
- Yoshida, T.; Sakuma, Y.; Todaka, D.; Maruyama, K.; Qin, F.; Mizoi, J.; Kidokoro, S.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of an Arabidopsis heat-shock transcription factor HsfA3 in the transcriptional cascade downstream of the DREB2A stress-regulatory system. Biochem. Biophys. Res. Commun. 2008, 368, 515–521. [Google Scholar] [PubMed]
- Qiu, Y.; Yu, D. Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ. Exp. Bot. 2009, 65, 35–47. [Google Scholar]
- Raineri, J.; Ribichich, K.F.; Chan, R.L. The sunflower transcription factor HaWRKY76 confers drought and flood tolerance to Arabidopsis thaliana plants without yield penalty. Plant Cell Rep. 2015, 34, 2065–2080. [Google Scholar] [PubMed]
- Wang, C.-T.; Ru, J.-N.; Liu, Y.-W.; Yang, J.-F.; Li, M.; Xu, Z.-S.; Fu, J.-D. The Maize WRKY Transcription Factor ZmWRKY40 Confers Drought Resistance in Transgenic Arabidopsis. Int. J. Mol. Sci. 2018, 19, 2580. [Google Scholar]
- Wang, C.-T.; Ru, J.-N.; Liu, Y.-W.; Li, M.; Zhao, D.; Yang, J.-F.; Fu, J.-D.; Xu, Z.-S. Maize WRKY Transcription Factor ZmWRKY106 Confers Drought and Heat Tolerance in Transgenic Plants. Int. J. Mol. Sci. 2018, 19, 3046. [Google Scholar]
- Huo, T.; Wang, C.-T.; Yu, T.-F.; Wang, D.-M.; Li, M.; Zhao, D.; Li, X.-T.; Fu, J.-D.; Xu, Z.-S.; Song, X.-Y. Overexpression of ZmWRKY65 transcription factor from maize confers stress resistances in transgenic Arabidopsis. Sci. Rep. 2021, 11, 4024. [Google Scholar]
- Sawa, M.; Nusinow, D.A.; Kay, S.A.; Imaizumi, T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 2007, 318, 261–265. [Google Scholar]
- Baek, D.; Kim, W.-Y.; Cha, J.-Y.; Park, H.J.; Shin, G.; Park, J.; Lim, C.J.; Chun, H.J.; Li, N.; Kim, D.H.; et al. The GIGANTEA-ENHANCED EM LEVEL Complex Enhances Drought Tolerance via Regulation of Abscisic Acid Synthesis. Plant Physiol. 2020, 184, 443–458. [Google Scholar]
- Sofo, A.; Scopa, A.; Nuzzaci, M.; Vitti, A. Ascorbate Peroxidase and Catalase Activities and Their Genetic Regulation in Plants Subjected to Drought and Salinity Stresses. Int. J. Mol. Sci. 2015, 16, 13561–13578. [Google Scholar] [CrossRef]
- Rossel, J.B.; Walter, P.B.; Hendrickson, L.; Chow, W.S.; Poole, A.; Mullineaux, P.M.; Pogson, B.J. A mutation affecting ASCORBATE PEROXIDASE 2 gene expression reveals a link between responses to high light and drought tolerance. Plant. Cell Environ. 2006, 29, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, Y.; Rao, J.; Wang, G.; Li, H.; Ge, F.; Chen, C. Overexpression of the glutathione S-transferase gene from Pyrus pyrifolia fruit improves tolerance to abiotic stress in transgenic tobacco plants. Mol. Biol. 2013, 47, 515–523. [Google Scholar] [CrossRef]
- Boisvert, S.; Laviolette, F.; Corbeil, J. Ray: Simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 2010, 17, 1519–1533. [Google Scholar] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Casimiro-Soriguer, C.S.; Muñoz-Mérida, A.; Pérez-Pulido, A.J. Sma3s: A universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics 2017, 17, 1700071. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef]
- Gómez-Gálvez, I.; Sánchez-Lucas, R.; San-Eufrasio, B.; Rodríguez de Francisco, L.E.; Maldonado-Alconada, A.M.; Fuentes-Almagro, C.; Castillejo, M.A. Optimizing shotgun proteomics analysis for a confident protein identification and quantitation in orphan plant species: The case of Holm oak (Quercus ilex). In Plant Proteomics. Methods in Molecular Biology, Vol. 2139; Jorrin-Novo, J., Valledor, L., Castillejo, M.A., Rey, M., Eds.; Springer: New York, NY, USA, 2020; pp. 157–168. ISBN 978-1-0716-0527-1. [Google Scholar]
- Silva, J.C.; Gorenstein, M.V.; Li, G.-Z.; Vissers, J.P.C.; Geromanos, S.J. Absolute Quantification of Proteins by LCMSE: A Virtue of Parallel ms Acquisition *S. Mol. Cell. Proteomics 2006, 5, 144–156. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar]
- Valledor, L.; Escandón, M.; Meijón, M.; Nukarinen, E.; Cañal, M.J.; Weckwerth, W. A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant J. 2014, 79, 173–180. [Google Scholar] [PubMed]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar]
- Shannon, P.; Markiel, A.; Owen, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Sánchez, V.M.; López-Hidalgo, C.; Rey, M.-D.; Castillejo, M.Á.; Jorrín-Novo, J.V.; Escandón, M. Multiomic Data Integration in the Analysis of Drought-Responsive Mechanisms in Quercus ilex Seedlings. Plants 2022, 11, 3067. https://doi.org/10.3390/plants11223067
Guerrero-Sánchez VM, López-Hidalgo C, Rey M-D, Castillejo MÁ, Jorrín-Novo JV, Escandón M. Multiomic Data Integration in the Analysis of Drought-Responsive Mechanisms in Quercus ilex Seedlings. Plants. 2022; 11(22):3067. https://doi.org/10.3390/plants11223067
Chicago/Turabian StyleGuerrero-Sánchez, Víctor M., Cristina López-Hidalgo, María-Dolores Rey, María Ángeles Castillejo, Jesús V. Jorrín-Novo, and Mónica Escandón. 2022. "Multiomic Data Integration in the Analysis of Drought-Responsive Mechanisms in Quercus ilex Seedlings" Plants 11, no. 22: 3067. https://doi.org/10.3390/plants11223067
APA StyleGuerrero-Sánchez, V. M., López-Hidalgo, C., Rey, M.-D., Castillejo, M. Á., Jorrín-Novo, J. V., & Escandón, M. (2022). Multiomic Data Integration in the Analysis of Drought-Responsive Mechanisms in Quercus ilex Seedlings. Plants, 11(22), 3067. https://doi.org/10.3390/plants11223067