

Genetic Dissection of Epistatic Interactions Contributing Yield-Related Agronomic Traits in Rice Using the Compressed Mixed Model

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Rice Datasets
2.2. Dimensionality Reduction
2.3. Genome-Wide Association Study
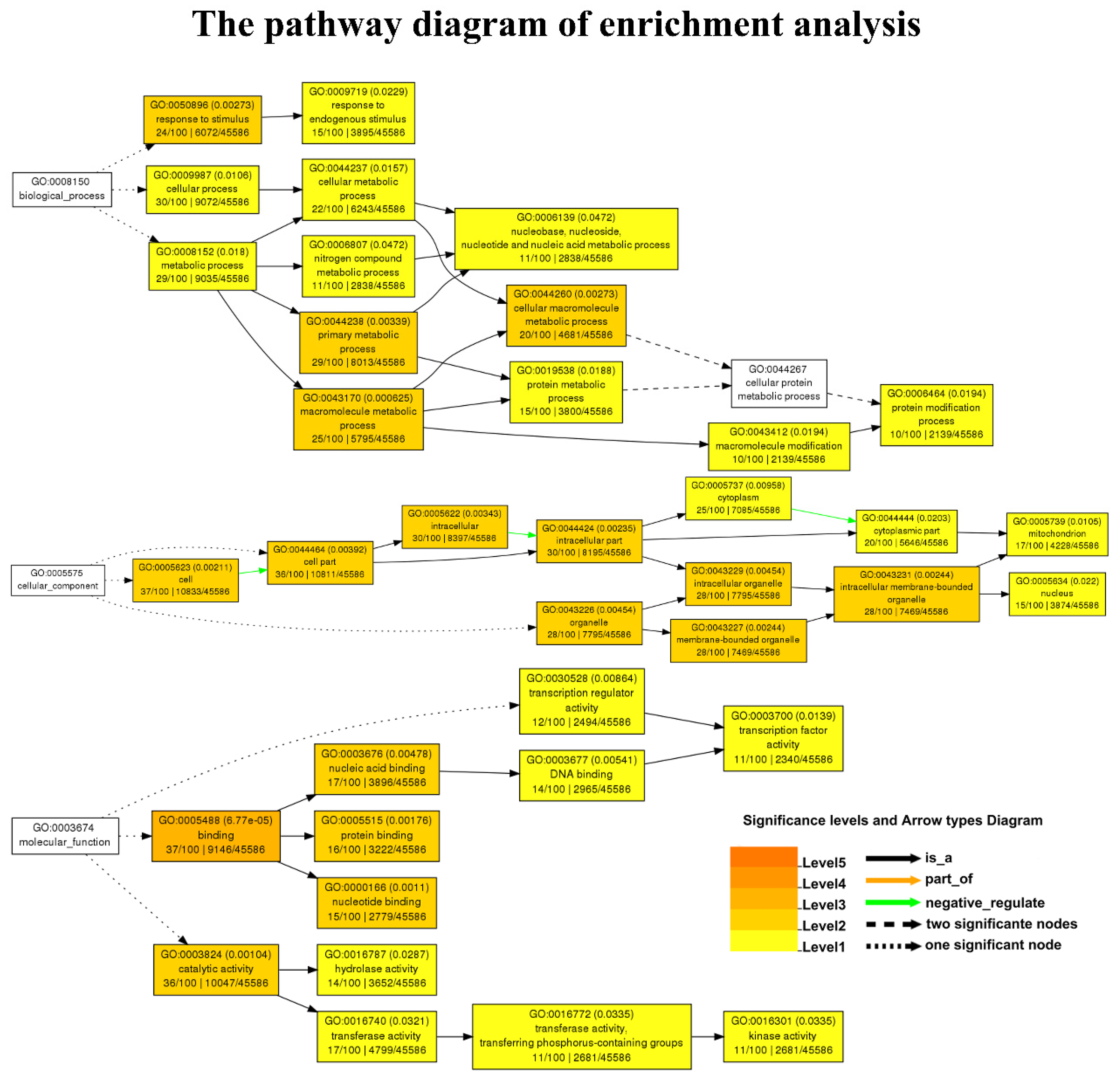
2.4. Candidate Gene Identification and Enrichment Analysis
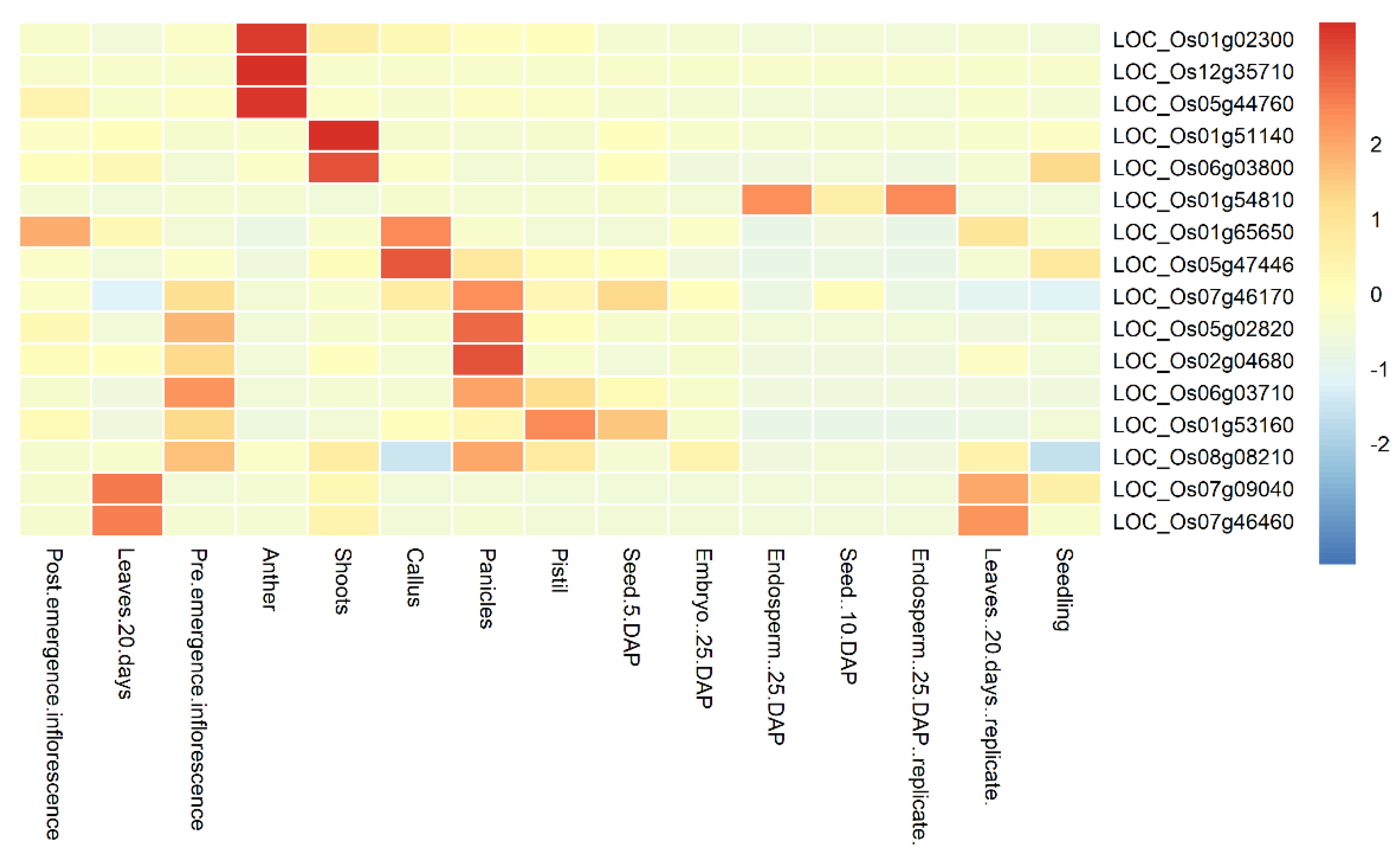
2.5. Tissue-Specific Expression Analysis
3. Results
3.1. Phenotypic Variation
3.2. Genetic Dissection of Epistatic Interactions
3.3. Functional Enrichment Analysis of the Candidate Genes
3.4. Expression Profile of the Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Butardo, V.M.; Sreenivasulu, N. Improving Head Rice Yield and Milling Quality: State-of-the-Art and Future Prospects. Methods Mol. Biol. 2019, 1892, 1–18. [Google Scholar] [PubMed]
- Sakamoto, T.; Matsuoka, M. Identifying and exploiting grain yield genes in rice. Curr. Opin. Plant Biol. 2008, 11, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Jiang, L.; Zheng, J.; Wang, T.; Wang, H.; Huang, Y.; Hong, Z. Genetic bases of rice grain shape: So many genes, so little known. Trends Plant Sci. 2013, 18, 218–226. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Liakat, A.M.; Price, A.H.; Norton, G.J.; Rafiqul, I.M.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2011, 44, 32–39. [Google Scholar] [CrossRef]
- Li, Z.; Pinson, S.R.; Park, W.D.; Paterson, A.H.; Stansel, J.W. Epistasis for three grain yield components in rice (Oryza sativa L.). Genetics 1997, 145, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. XV.—The Correlation between Relatives on the Supposition of Mendelian Inheritance. Trans. R. Soc. Edinb. 1919, 52, 399–433. [Google Scholar] [CrossRef]
- Carlborg, O.; Haley, C.S. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 2004, 5, 618–625. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Zhang, Z.; Xiang, Y.; Liu, M.; Zhou, Y.; Zuo, J.; Zhang, H.; Chen, Y.; Zhang, Y. A compressed variance component mixed model for detecting QTNs, and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 2022, 15, 630–650. [Google Scholar] [CrossRef]
- Chang, Y.C.; Wu, J.T.; Hong, M.Y.; Tung, Y.A.; Hsieh, P.H.; Yee, S.W.; Giacomini, K.M.; Oyang, Y.J.; Chen, C.Y.; Weiner, M.W.; et al. GenEpi: Gene-based epistasis discovery using machine learning. BMC Bioinform. 2020, 21, 68. [Google Scholar] [CrossRef]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Brown, K.T.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.I.W.D.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, Z.; Wan, X.; Yang, Q.; Xue, H.; Yu, W. SNPHarvester: A filtering-based approach for detecting epistatic interactions in genome-wide association studies. Bioinformatics 2009, 25, 504–511. [Google Scholar] [CrossRef]
- Thierry, S.; Ioannis, X.; Sven, B.; Karen, K. FastEpistasis: A high performance computing solution for quantitative trait epistasis. Bioinformatics 2010, 26, 1468–1469. [Google Scholar]
- Wan, X.; Yang, C.; Yang, Q.; Xue, H.; Fan, X.; Tang, N.L.S.; Yu, W. BOOST: A Fast Approach to Detecting Gene-Gene Interactions in Genome-wide Case-Control Studies. Am. J. Hum. Genet. 2010, 87, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. New strategies for identifying gene-gene interactions in hypertension. Ann. Med. 2002, 34, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.F.; König, I.R.; Ziegler, A. On safari to Random Jungle: A fast implementation of Random Forests for high-dimensional data. Bioinformatics 2010, 26, 1752–1758. [Google Scholar] [CrossRef]
- Stephan, J.; Stegle, O.; Beyer, A. A random forest approach to capture genetic effects in the presence of population structure. Nat. Commun. 2015, 6, 7432. [Google Scholar] [CrossRef]
- Ma, L.; Brautbar, A.; Boerwinkle, E.; Sing, C.F.; Clark, A.G.; Keinan, A. Knowledge-driven analysis identifies a gene-gene interaction affecting high-density lipoprotein cholesterol levels in multi-ethnic populations. PLoS Genet. 2012, 8, e1002714. [Google Scholar] [CrossRef]
- Crawford, L.; Zeng, P.; Mukherjee, S.; Zhou, X. Detecting epistasis with the marginal epistasis test in genetic mapping studies of quantitative traits. PLoS Genet. 2017, 13, e1006869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Mao, Y.; Xie, C.; Smith, H.; Luo, L.; Xu, S. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 2005, 169, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh, B.I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ersoz, E.; Lai, C.-Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef]
- Xiang, Z.; Matthew, S. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. Editorial: The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Zhang, Y.-M. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, F.; Zhou, Y. The Application of Multi-Locus GWAS for the Detection of Salt-Tolerance Loci in Rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yang, Y.; Li, H.; Liu, Q.; Zhang, J.; Yin, J.; Chu, S.; Zhang, X.; Yu, K.; Lv, L.; et al. Genome-Wide Association Studies of Photosynthetic Traits Related to Phosphorus Efficiency in Soybean. Front. Plant Sci. 2018, 9, 1226. [Google Scholar]
- Moore, J.H.; Asselbergs, F.W.; Williams, S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010, 26, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Tan, Y.; Hua, J.P.; Sun, X.; Xu, C.; Zhang, Q. Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2002, 105, 248–257. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, H.; Zhang, G.; Li, L.; Ye, G. Dynamic analysis of QTLs on tiller number in rice (Oryza sativa L.) with single segment substitution lines. TAG. Theor. Appl. Genet. Theor. Und Angew. Genet. 2012, 125, 143–153. [Google Scholar] [CrossRef]
- Divya, B.; Malathi, S.; Rao, Y.V.; Raju, A.K.; Sukumar, M.; Kavitha, B.; Sarla, N. Detecting CSSLs and yield QTLs with additive, epistatic and QTL×environment interaction effects from Oryza sativa × O. nivara IRGC81832 cross. Sci. Rep. 2020, 10, 7766. [Google Scholar]
- Okada, S.; Iijima, K.; Hori, K.; Yamasaki, M. Genetic and epistatic effects for grain quality and yield of three grain-size QTLs identified in brewing rice (Oryza sativa L.). Mol. Breed. New Strateg. Plant Improv. 2020, 40, 88. [Google Scholar] [CrossRef]
- Ueki, M.; Cordell, H.J. Improved statistics for genome-wide interaction analysis. PLoS Genet. 2012, 8, e1002625. [Google Scholar] [CrossRef]
- Guo, X.; Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Genome-wide association analyses using a Bayesian approach for litter size and piglet mortality in Danish Landrace and Yorkshire pigs. BMC Genom. 2016, 17, 468. [Google Scholar] [CrossRef] [PubMed]
- Xu, S. An expectation-maximization algorithm for the Lasso estimation of quantitative trait locus effects. Heredity 2010, 105, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Li, G.; Zhang, Y.M. A compressed variance component mixed model framework for detecting small and linked QTL-by-environment interactions. Brief. Bioinform. 2022, 23, bbab596. [Google Scholar] [CrossRef]
- Zobaer, A.; Asif, A.M.; Munirul, A.; Haque, M.M.N. Robustification of GWAS to explore effective SNPs addressing the challenges of hidden population stratification and polygenic effects. Sci. Rep. 2021, 11, 13060. [Google Scholar]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.B.; Li, J.X.; Xu, C.G.; Tan, Y.F.; Li, X.H.; Zhang, Q. Identification of quantitative trait loci and epistatic interactions for plant height and heading date in rice. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2002, 104, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Dong, X.; Han, X.; Zhang, F.; Zhu, Y.; Xin, X.; Wang, Y.; Hu, Y.; Yuan, D.; Wang, J.; et al. OsPDCD5 negatively regulates plant architecture and grain yield in rice. Proc. Natl. Acad. Sci. USA 2021, 118, e2018799118. [Google Scholar] [CrossRef]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, P.; Zhang, H.; Meng, C.; Zhang, X.; Hou, J.; Wei, C. Low soil temperature reducing the yield of drip irrigated rice in arid area by influencing anther development and pollination. J. Arid Land 2019, 11, 419–430. [Google Scholar] [CrossRef]
- Ning, C.; Wang, D.; Kang, H.; Mrode, R.; Zhou, L.; Xu, S.; Liu, J.F. A rapid epistatic mixed-model association analysis by linear retransformations of genomic estimated values. Bioinformatics 2018, 34, 1817–1825. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NO. | QTN1 | QTN2 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Pos | Gene ID | Gene | Chr | Pos | Gene ID | Gene | LOD | aa.Effect | ad.Effect | da.Effect | dd.Effect | Variance | PVE (%) | p-Value | |
| QTN1 | 1 | 38111539 | 6.79 | 12.63 | −19.11 | 57.09 | 12.832 | 1.62 × 10−7 | ||||||||
| QQI 1 | 1 | 723562 | 11 | 25849860 | 5.75 | 3.76 | 12.94 | 2.910 | 2.66 × 10−7 | |||||||
| QQI 2 | 1 | 29384858 | 2 | 2233430 | LOC_Os02g04680 | LCRN1; OsSPL3 | 5.68 | 2.69 | 6.03 | 31.82 | 7.151 | 2.09 × 10−6 | ||||
| QQI 3 | 1 | 29557152 | 12 | 15325876 | 6.87 | 5.75 | 24.38 | 5.480 | 1.85 × 10−8 | |||||||
| QQI 4 | 1 | 30547272 | LOC_Os01g53160 | OFP3; OsOFP04 | 5 | 1015771 | 5.56 | −4.94 | 21.98 | 4.940 | 4.24 × 10−7 | |||||
| QQI 5 | 1 | 31651011 | LOC_Os01g54810 LOC_Os01g54930 | THIS1 OsVOZ1 | 9 | 9425939 | 3.23 | −4.19 | 6.25 | 1.406 | 1.14 × 10−4 | |||||
| QQI 6 | 1 | 39419765 | LOC_Os01g68000 | PLA2; LHD2 | 6 | 3636360 | 6.97 | 3.69 | 2.58 | 9.16 | 2.059 | 1.07 × 10−7 | ||||
| QQI 7 | 3 | 13773095 | LOC_Os03g24220 | VLN2 | 9 | 11706989 | 5.72 | 4.36 | 8.18 | 1.839 | 2.89 × 10−7 | |||||
| QQI 8 | 4 | 29851050 | 12 | 21716878 | 6.32 | −4.51 | 20.16 | 4.531 | 6.89 × 10−8 | |||||||
| QQI 9 | 5 | 25953209 | LOC_Os05g44310 LOC_Os05g44760 | OsSec18 OsHXK5 | 6 | 1524748 | LOC_Os06g03710 LOC_Os06g03770 LOC_Os06g03810 LOC_Os06g04010 | DLT; SMOS2 OsATM3 ROD1 OsGBP1 | 4.65 | 3.20 | 8.66 | 1.946 | 3.69 × 10−6 | |||
| QQI 10 | 5 | 27196868 | LOC_Os05g47446 | OsPDCD5 | 8 | 4731022 | LOC_Os08g08210 | SDG701 | 4.06 | 3.99 | 7.59 | 1.707 | 1.52 × 10−5 | |||
| QQI 11 | 5 | 28309324 | 7 | 26640298 | 4.18 | −4.67 | −3.22 | 14.83 | 3.334 | 6.67 × 10−5 | ||||||
| QQI 12 | 6 | 29357275 | LOC_Os06g48530 | Du13 | 7 | 6016810 | 5.88 | 2.54 | 5.07 | 1.139 | 1.95 × 10−7 | |||||
| QQI 13 | 7 | 4724800 | 7 | 27550702 | LOC_Os07g46460 | Fd-GOGAT1 | 4.79 | 5.73 | −1.32 | 22.64 | 5.090 | 1.61 × 10−5 | ||||
| QQI 14 | 8 | 51045 | 9 | 854638 | 8.16 | 4.86 | 15.52 | 3.488 | 8.76 × 10−10 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Wu, X.; Chen, J.; Wang, S.; Wan, Y.; Ji, H.; Wen, Y.; Zhang, J. Genetic Dissection of Epistatic Interactions Contributing Yield-Related Agronomic Traits in Rice Using the Compressed Mixed Model. Plants 2022, 11, 2504. https://doi.org/10.3390/plants11192504
Li L, Wu X, Chen J, Wang S, Wan Y, Ji H, Wen Y, Zhang J. Genetic Dissection of Epistatic Interactions Contributing Yield-Related Agronomic Traits in Rice Using the Compressed Mixed Model. Plants. 2022; 11(19):2504. https://doi.org/10.3390/plants11192504
Chicago/Turabian StyleLi, Ling, Xinyi Wu, Juncong Chen, Shengmeng Wang, Yuxuan Wan, Hanbing Ji, Yangjun Wen, and Jin Zhang. 2022. "Genetic Dissection of Epistatic Interactions Contributing Yield-Related Agronomic Traits in Rice Using the Compressed Mixed Model" Plants 11, no. 19: 2504. https://doi.org/10.3390/plants11192504
APA StyleLi, L., Wu, X., Chen, J., Wang, S., Wan, Y., Ji, H., Wen, Y., & Zhang, J. (2022). Genetic Dissection of Epistatic Interactions Contributing Yield-Related Agronomic Traits in Rice Using the Compressed Mixed Model. Plants, 11(19), 2504. https://doi.org/10.3390/plants11192504