
Establishing MinION Sequencing and Genome Assembly Procedures for the Analysis of the Rooibos (Aspalathus linearis) Genome

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Purification and Quantification
2.2. Genome Sequencing
2.3. Genome Assemblies and Evaluation
3. Results
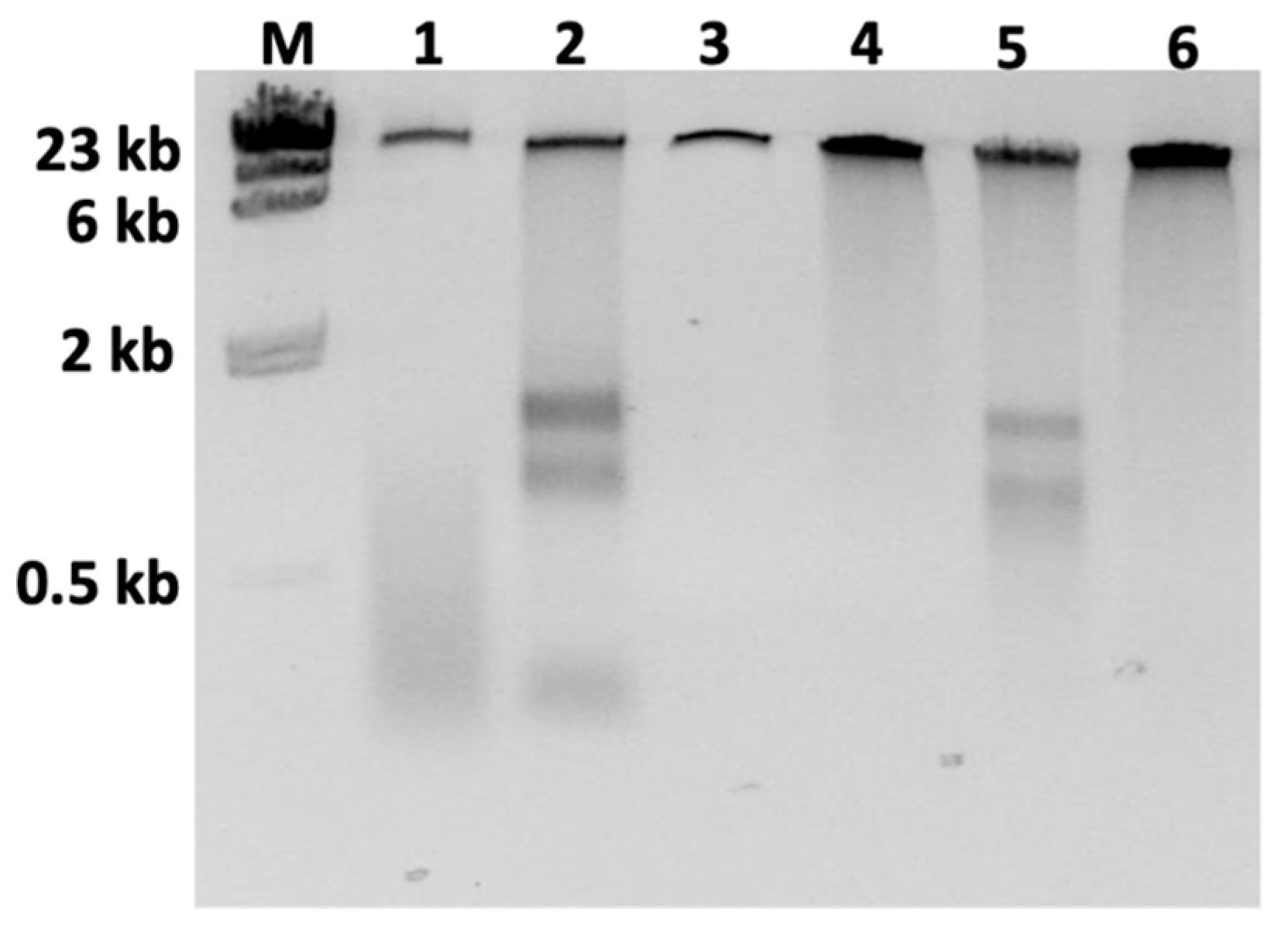
3.1. Optimizing DNA Sample Purity
3.2. Nanopore Sequencing
3.3. Evaluation of Genome Assemblies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dumschott, K.; Schmidt, M.H.W.; Chawla, H.S.; Snowdon, R.; Usadel, B. Oxford Nanopore sequencing: New opportunities for plant genomics? J. Exp. Bot. 2020, 71, 5313–5322. [Google Scholar] [CrossRef]
- Murigneux, V.; Rai, S.K.; Furtado, A.; Bruxner, T.J.C.; Tian, W.; Harliwong, I.; Wei, H.; Yang, B.; Ye, Q.; Anderson, E.; et al. Comparison of long-read methods for sequencing and assembly of a plant genome. Gigascience 2020, 9, 146. [Google Scholar] [CrossRef]
- Pushkova, E.N.; Beniaminov, A.D.; Krasnov, G.S.; Novakovskiy, R.O.; Povkhova, L.V.; Melnikova, N.V.; Dmitriev, A.A. Extraction of high-molecular-weight DNA from poplar plants for Nanopore sequencing. Plant Biotechnol. Postgenom. Era 2019, 158–160. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Building near-complete plant genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Torkel, C.; Stanley, D.; Nasim, J.; Borevitz, J.; Schwessinger, B. High-molecular weight DNA extraction, clean-up and size selection for long-read sequencing. PLoS ONE 2021, 16, e0253830. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Maaty, N.A.-F.; Oraby, H.A.-S. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. Natl. Res. Cent. 2019, 43, 25. [Google Scholar] [CrossRef]
- Stander, E.A.; Williams, W.; Rautenbach, F.; Roes-Hill, M.L.; Mgwatyu, Y.; Marnewick, J.; Hesse, U. Visualization of Aspalathin in Rooibos (Aspalathus linearis) Plant and Herbal Tea Extracts Using Thin-Layer Chromatography. Molecules 2019, 24, 938. [Google Scholar] [CrossRef] [PubMed]
- Stander, M.A.; van Wyk, B.E.; Taylor, M.J.C.; Long, H.S. Analysis of Phenolic Compounds in Rooibos Tea (Aspalathus linearis) with a Comparison of Flavonoid-Based Compounds in Natural Populations of Plants from Different Regions. J. Agric. Food Chem. 2017, 65, 10270–10281. [Google Scholar] [CrossRef]
- Krafczyk, N.; Glomb, M.A. Characterization of phenolic compounds in rooibos tea. J. Agric. Food Chem. 2008, 56, 3368–3376. [Google Scholar] [CrossRef]
- Dludla, P.v.; Muller, C.J.F.; Louw, J.; Mazibuko-Mbeje, S.E.; Tiano, L.; Silvestri, S.; Orlando, P.; Marcheggiani, F.; Cirilli, I.; Chellan, N.; et al. The combination effect of aspalathin and phenylpyruvic acid-2-o-β-d-glucoside from rooibos against hyperglycemia-induced cardiac damage: An in vitro study. Nutrients 2020, 12, 1151. [Google Scholar] [CrossRef]
- Dludla, P.V.; Joubert, E.; Louw, J.; Muller, C.C.J.; Huisamen, B.; Essop, M.F.; Johnson, R. A phenylpropenoic acid glucoside (PPAG) of Aspalathus linearis protects H9c2 cardiomyocytes against hyperglycemia-induced cell apoptosis. Planta Med. 2015, 81, PM_195. [Google Scholar] [CrossRef]
- Stander, E.A.; Williams, W.; Mgwatyu, Y.; Heusden, P.v.; Rautenbach, F.; Marnewick, J.; Roes-Hill, M.L.; Hesse, U. Transcriptomics of the rooibos (Aspalathus linearis) species complex. BioTech 2020, 9, 19. [Google Scholar] [CrossRef]
- Mgwatyu, Y.; Stander, A.A.; Ferreira, S.; Williams, W.; Hesse, U. Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses. Plants 2020, 9, 270. [Google Scholar] [CrossRef]
- Stander, A. De Novo Assembly of the Rooibos Genome. Master's Thesis, University of the Western Cape, Cape Town, South Africa, 2020. Available online: http://etd.uwc.ac.za/xmlui/handle/11394/7719 (accessed on 25 June 2022).
- Yang, T.; Wu, C. DNA Extraction for Plant Samples by CTAB; GigaScience Press: Shenzen, China, 2018. [Google Scholar]
- Sample & Assay Technologies QIAGEN® Genomic DNA Handbook 2005. Available online: www.qiagen.com (accessed on 23 May 2022).
- Community-Knowledge-Extraction methods-@extraction_method.title.humanize. Available online: https://community.nanoporetech.com/extraction_methods/arabidopsis-leaf-dna (accessed on 25 June 2022).
- Introducing FilterByTile: Remove Low-Quality Reads without Adding Bias. Available online: https://www.biostars.org/p/228762/ (accessed on 25 June 2022).
- Babraham Bioinformatics-FastQC. A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 June 2022).
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef]
- de Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef]
- Zimin, A.v.; Marçais, G.; Puiu, D.; Roberts, M.; SSalzberg, L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef]
- Haghshenas, E.; Asghari, H.; Stoye, J.; Chauve, C.; Hach, F. HASLR: Fast Hybrid Assembly of Long Reads. IScience 2020, 23, 101389. [Google Scholar] [CrossRef] [PubMed]
- di Genova, A.; Buena-Atienza, E.; Ossowski, S.; Sagot, M.F. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 2020, 39, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Šikić, M. Time- and memory-efficient genome assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking long-read assemblers for genomic analyses of bacterial pathogens using oxford nanopore sequencing. Int. J. Mol. Sci. 2020, 21, 9161. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; APhillippy, M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Nextomics/NextDenovo: Fast and Accurate De Novo Assembler for Long Reads. Available online: https://github.com/Nextomics/NextDenovo (accessed on 5 August 2022).
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kong, M.; Oh, J.; Lim, J.S.; Chung, S.H.; Kim, J.-M.; Kim, J.-S.; Kim, K.-H.; Yoo, J.-C.; Kwak, W. Comparative evaluation of Nanopore polishing tools for microbial genome assembly and polishing strategies for downstream analysis. Sci. Rep. 2021, 11, 20740. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Polishing the Oxford Nanopore long-read assemblies of bacterial pathogens with Illumina short reads to improve genomic analyses. Genomics 2021, 113, 1366–1377. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Sivakumaran, K.; Amarakoon, S.; Sivakumaran, C.K. Bioactivity of fruit teas and tisanes—A review. J. Pharmacogn. Phytochem. 2018, 7, 323–327. [Google Scholar]
- Scaldaferri, M.M.; Freitas, J.S.; Santos, E.S.L.; Vieira, J.G.P.; Gonçalves, Z.S.; Cerqueira-Silva, C.B.M. Comparison of protocols for genomic DNA extraction from ‘velame pimenta’ (Croton linearifolius), a native species to the Caatinga, Brazil. Afr. J. Biotechnol. 2016, 12, 4761–4766. [Google Scholar] [CrossRef]
- Abubakar, B.M.; Izham, N.H.M.; Salleh, F.M.; Omar, M.S.S.; Wagiran, A. Comparison of Different DNA Extraction Methods from Leaves and Roots of Eurycoma longifolia Plant. Adv. Sci. Lett. 2018, 24, 3641–3645. [Google Scholar] [CrossRef]
- Esfandani-Bozchaloyi, S.; Sheidai, M.; Kalalegh, M.H. Comparison of DNA extraction methods from Geranium (Geraniaceae). Acta Bot. Hung. 2019, 61, 251–266. [Google Scholar] [CrossRef]
- Dehestani, A.; Tabar, S.K.K. A rapid efficient method for DNA isolation from plants with high levels of secondary metabolites. Asian J. Plant Sci. 2007, 6, 977–981. [Google Scholar] [CrossRef][Green Version]
- Schalamun, M.; Nagar, R.; Kainer, D.; Beavan, E.; Eccles, D.; Rathjen, J.P.; Lanfear, R.; Schwessinger, B. Harnessing the MinION: An example of how to establish long-read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Mol. Ecol. Resour. 2019, 19, 77–89. [Google Scholar] [CrossRef]
- Obtaining High Quality DNA from Plant Tissues for Nanopore Sequencing|Stella Loke—YouTube. Available online: https://www.youtube.com/watch?v=acaFw9mHGVw&t=304s (accessed on 25 June 2022).
- “ZymocleanTM Large Fragment DNA Recovery Kit”. Available online: https://files.zymoresearch.com/protocols/_d4001t_d4001_d4002_d4007_d4008_zymoclean_gel_dna_recovery_kit.pdf (accessed on 25 June 2022).
- Community-Knowledge-Extraction Methods-Plant Leaf Gdna. Available online: https://community.nanoporetech.com/extraction_method_groups/plant-leaf-gDNA (accessed on 25 June 2022).
- Vaillancourt, B.; Buell, C.R. High molecular weight DNA isolation method from diverse plant species for use with Oxford Nanopore sequencing. BioRxiv 2019, 783159. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Pushkova, E.N.; Novakovskiy, R.O.; Beniaminov, A.D.; Rozhmina, T.A.; Zhuchenko, A.A.; Bolsheva, N.L.; Muravenko, O.V.; Povkhova, L.V.; Dvorianinova, E.M.; et al. Genome Sequencing of Fiber Flax Cultivar Atlant Using Oxford Nanopore and Illumina Platforms. Front. Genet. 2021, 11, 590282. [Google Scholar] [CrossRef]
- Driguez, P.; Bougouffa, S.; Carty, K.; Putra, A.; Jabbari, K.; Reddy, M.; Soppe, R.; Cheung, M.S.; Fukasawa, Y.; Ermini, L. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome Biol. 2021, 22, 256. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, R.; Chen, C.; Sigwart, J.D.; Kocot, K.M. Benchmarking Oxford Nanopore read assemblers for high-quality molluscan genomes. Philos. Trans. R. Soc. B 2021, 376, 20200160. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Das, A.; Kainer, D.; Schalamun, M.; Morales-Suarez, A.; Schwessinger, B.; Lanfear, R. The draft nuclear genome assembly of Eucalyptus pauciflora: A pipeline for comparing de novo assemblies. Gigascience 2020, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Jeon, M.S.; Hodgett, M.; Waterhouse, P.; Eyun, S.i. Comparative Evaluation of Genome Assemblers from Long-Read Sequencing for Plants and Crops. J. Agric. Food Chem. 2020, 68, 7670–7677. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Choi, S.C.; Kang, Y.; Kim, K.M.; Kang, C.S.; Kim, C. Constructing a reference genome in a single lab: The possibility to use oxford nanopore technology. Plants 2019, 8, 270. [Google Scholar] [CrossRef]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef]


{kind=link}
| DNA Extraction Method | Plant | Purification Method + | 260/280 | 260/230 | Qubit (ng/µL) | Nanodrop (ng/µL) | Q:ND * | Starting Material | Total Yield (µg) | Sequencing Run |
|---|---|---|---|---|---|---|---|---|---|---|
| SDS | 1 | C:I extraction | 2.12 | 1.93 | 20.8 | 67.5 | 0.3 | 1 g leaves | 2.0 | n/a |
| CTAB | 1 | None | 2.11 | 2.01 | 358 | 1944 | 0.2 | 1 g leaves | 35.8 | 1 |
| CTAB | 1 | Zymoclean | 2.94 | 0.04 | 6.3 | 13.0 | 0.5 | 3.5 µg DNA | 0.06 | n/a |
| CTAB | 1 | Genomic-tip 1 | 2.07 | 1.91 | 102 | 1816 | 0.1 | 100 µg DNA | 2.0 | 3 |
| CTAB | 1 | Genomic-tip 2 | 2.06 | 2.22 | 347 | 1143 | 0.3 | 100 µg DNA | 6.9 | 4 |
| CTAB | 1 | DNeasy | 1.92 | 2.38 | 323 | 337 | 1.0 | 10 µg DNA | 6.4 | 2 |
| CTAB | 2 | DNeasy | 1.98 | 2.35 | 222 | 260 | 0.9 | 10 µg DNA | 4.4 | 6 |
| CTAB | 1 | DNeasy | 1.92 | 2.33 | 325 | 381 | 0.9 | 10 µg DNA | 6.5 | 7 |
| CTAB | 1 | DNeasy | 1.97 | 2.35 | 324 | 336 | 1.0 | 10 µg DNA | 6.4 | 8 |
| Run1 | Run2 | Run3 | Run4 | Run5 | Run6 | Run7 | |
|---|---|---|---|---|---|---|---|
| total gigabases (Gbp) | 1.01 | 4.56 | 6.62 | 19.08 | 11.24 | 4.26 | 2.24 |
| # reads (×1 M) | 0.30 | 1.21 | 1.56 | 4.98 | 3.91 | 4.25 | 2.30 |
| N50 length (bp) | 4372 | 6002 | 6641 | 6483 | 5642 | 1666 | 1480 |
| median length (bp) | 2656 | 2519 | 3027 | 2496 | 1607 | 569 | 587 |
| max length (bp) | 60,435 | 11,5278 | 75,142 | 79,588 | 180,920 | 116,028 | 110,672 |
| median Quality | 10 | 11.1 | 11.4 | 11.3 | 11.2 | 11.2 | 11 |
| Read length distributions: | |||||||
| >10 kb | 11,270 | 86,876 | 136,418 | 401,054 | 167,363 | 10,551 | 4616 |
| >20 kb | 598 | 13,964 | 17,790 | 54,087 | 6700 | 460 | 215 |
| >50 kb | 2 | 91 | 61 | 123 | 2 | 3 | 4 |
| >100 kb | 0 | 1 | 0 | 0 | 1 | 1 | 2 |
| >200 kb | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Top 5 longest reads (bp) and their mean quality score: | |||||||
| 1 | 60,435 (9.0) | 115,278 (10.0) | 75,142 (10.9) | 79,588 (8.5) | 180,920 (19.6) | 116,028 (7.9) | 110,672 (14.0) |
| 2 | 57,561 (8.8) | 84,582 (9.8) | 71,014 (9.0) | 75,403 (11.6) | 135,178 (7.1) | 64,587 (14.9) | 102,055 (14.7) |
| 3 | 45,472 (10.8) | 74,614 (12.7) | 69,281 (10.6) | 74,146 (13.1) | 91,826 (7.9) | 61,155 (7.7) | 99,741 (6.7) |
| 4 | 42,770 (9.6) | 71,567 (10.0) | 65,014 (11.0) | 70,447 (9.3) | 47,153 (10.2) | 44,661 (10.3) | 95,441 (14.8) |
| 5 | 42,761 (9.6) | 71,536 (11.0) | 64,036 (10.0) | 68,961 (10.1) | 46,147 (12.4) | 39,799 (9.9) | 90,125 (14.6) |
| Assembly Parameters | Illumina | Illumina + MinION | |||
|---|---|---|---|---|---|
| Platanus | MaSuRCA | MaSuRCA | Haslr | Wengan | |
| Number of scaffolds | 78,315 | 70,462 | 29,263 | 19,723 | 12,972 |
| Largest scaffold (bp) | 154,059 | 257,349 | 1,225,954 | 77,419 | 222,008 |
| Total length (Mbp) | 693.7 | 857.9 | 1482.6 | 173.6 | 218.6 |
| N50 length (bp) | 10,871 | 17,142 | 80,888 | 10,950 | 22,199 |
| Complete BUSCOs (%) | 56.1 | 45.5 | 84.3 | 67.0 | 49.4 |
| Complete single BUSCOs (%) | 51.8 | 36.9 | 49.0 | 62.7 | 48.6 |
| Complete duplicated BUSCOs (%) | 4.3 | 5.9 | 35.3 | 4.3 | 0.8 |
| Fragmented BUSCOs (%) | 34.5 | 18.8 | 2.0 | 12.9 | 8.6 |
| Missing BUSCOs (%) | 9.4 | 35.7 | 13.7 | 20.1 | 42.0 |
| Assembly Parameters | Unpolished | Racon_Long | Medaka | Racon_Short | Nextpolish | |
|---|---|---|---|---|---|---|
| Flye | Number of contigs | 33,346 | 32,286 | 32,393 | 31,921 | 32,392 |
| Largest contig (bp) | 921,299 | 925,383 | 928,488 | 926,118 | 925,075 | |
| Total length (Mbp) | 1118.5 | 1107.2 | 1112.5 | 1107.4 | 1110.7 | |
| N50 length (bp) | 77,709 | 78,172 | 78,358 | 78,512 | 78,176 | |
| Complete BUSCOs (%) | 97.3 | 96.5 | 98.0 | 99.2 | 99.2 | |
| Complete single BUSCOs (%) | 65.9 | 71.8 | 70.2 | 65.5 | 64.3 | |
| Complete duplicated BUSCOs (%) | 31.4 | 24.7 | 27.8 | 33.7 | 34.9 | |
| Fragmented BUSCOs (%) | 2.4 | 2.7 | 1.6 | 0.8 | 0.8 | |
| Missing BUSCOs (%) | 0.3 | 0.8 | 0.4 | 0.0 | 0.0 | |
| Canu | Number of contigs | 33,477 | 33,351 | 33,357 | 33,320 | 33,356 |
| Largest contig (bp) | 434,840 | 443,413 | 444,838 | 443,863 | 443,487 | |
| Total length (Mbp) | 949.0 | 965.0 | 970.8 | 968.1 | 969.1 | |
| N50 length (bp) | 40,175 | 41,134 | 41,324 | 41,225 | 41,247 | |
| Complete BUSCOs (%) | 96.1 | 98.8 | 99.2 | 99.6 | 99.6 | |
| Complete single BUSCOs (%) | 71.4 | 72.5 | 72.5 | 57.6 | 56.9 | |
| Complete duplicated BUSCOs (%) | 24.7 | 26.3 | 26.7 | 42.0 | 42.7 | |
| Fragmented BUSCOs (%) | 2.4 | 0.0 | 0.4 | 0.0 | 0.0 | |
| Missing BUSCOs (%) | 1.5 | 1.2 | 0.4 | 0.4 | 0.4 | |
| Raven | Number of contigs | 11,675 | 11,674 | 11,674 | 11,672 | 11,674 |
| Largest contig (bp) | 760,847 | 765,889 | 767,221 | 765,347 | 764,913 | |
| Total length (Mbp) | 905.6 | 909.8 | 915.2 | 912.6 | 913.1 | |
| N50 length (bp) | 99,352 | 99,903 | 100,315 | 100,069 | 100,060 | |
| Complete BUSCOs (%) | 96.9 | 95.7 | 96.9 | 97.6 | 98.0 | |
| Complete single BUSCOs (%) | 79.6 | 76.9 | 79.6 | 69.0 | 69.4 | |
| Complete duplicated BUSCOs (%) | 17.3 | 18.8 | 17.3 | 28.6 | 28.6 | |
| Fragmented BUSCOs (%) | 1.2 | 2.4 | 1.2 | 0.8 | 0.8 | |
| Missing BUSCOs (%) | 1.9 | 1.9 | 1.9 | 1.6 | 1.2 | |
| Redbean | Number of contigs | 16,753 | 16,347 | 16,350 | 16,275 | 16,350 |
| Largest contig (bp) | 1,686,036 | 1,739,027 | 1,738,674 | 1,734,655 | 1,734,062 | |
| Total length (Mbp) | 962.0 | 993.5 | 996.8 | 994.0 | 995.1 | |
| N50 length (bp) | 142,771 | 148,572 | 148,398 | 148,157 | 148,018 | |
| Complete BUSCOs (%) | 83.9 | 96.5 | 98.8 | 99.2 | 99.2 | |
| Complete single BUSCOs (%) | 80.4 | 82.4 | 85.9 | 74.9 | 75.3 | |
| Complete duplicated BUSCOs (%) | 3.5 | 14.1 | 12.9 | 24.3 | 23.9 | |
| Fragmented BUSCOs (%) | 8.6 | 2.7 | 0.4 | 0.4 | 0.4 | |
| Missing BUSCOs (%) | 7.5 | 0.8 | 0.8 | 0.4 | 0.4 | |
| NextDenovo | Number of contigs | 5431 | 5431 | 5431 | 5431 | 5431 |
| Largest contig (bp) | 3,406,093 | 3,359,923 | 3,371,738 | 3,361,759 | 3,361,805 | |
| Total length (Mbp) | 824.2 | 817.1 | 820.9 | 818.6 | 818.9 | |
| N50 length (bp) | 218,190 | 217,600 | 218,885 | 218,094 | 218,285 | |
| Complete BUSCOs (%) | 90.2 | 92.1 | 93.7 | 94.5 | 94.5 | |
| Complete single BUSCOs (%) | 72.9 | 72.5 | 76.1 | 69.8 | 69.8 | |
| Complete duplicated BUSCOs (%) | 17.3 | 19.6 | 17.6 | 24.7 | 24.7 | |
| Fragmented BUSCOs (%) | 3.1 | 1.2 | 0.4 | 0.4 | 0.4 | |
| Missing BUSCOs (%) | 6.7 | 6.7 | 5.9 | 5.1 | 5.1 |
| Program | Data | Cores | CPU Time (h) | RAM Memory (GB) | Disc Space (GB) |
|---|---|---|---|---|---|
| Platanus | Illumina | 56 | 1308 | 624 | 5 |
| MaSuRCA | Illumina | 56 | 1420 | 426 | 245 |
| Haslr | Illumina + MinION | 56 | 37 | 39 | 50 |
| Wengan | Illumina + MinION | 56 | 465 | 599 | 37 |
| MaSuRCA | Illumina + MinION | 56 | 2462 | 864 | 543 |
| Flye | MinION | 56 | 863 | 584 | 205 |
| Raven | MinION | 56 | 44 | 36 | 24 |
| Redbean | MinION | 56 | 205 | 54 | 16 |
| Canu | MinION | 112 | 35,401 | 2960 | 57 |
| NextDenovo | MinION | 56 | 244 | 804 | 7.2 |
| Racon_long * | Polishing | 56 | 186 ± 1.5 | 776 ± 0.0 | 577 ± 0.0 |
| Medaka * | Polishing | 56 | 33 ± 0.0 | 106 ± 0.1 | 40 ± 0.0 |
| Racon_short * | Polishing | 56 | 167 ± 0.3 | 654 ± 0.0 | 554 ± 0.0 |
| Nextpolish * | Polishing | 56 | 37 ± 0.5 | 54 ± 0.0 | 51 ± 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mgwatyu, Y.; Cornelissen, S.; van Heusden, P.; Stander, A.; Ranketse, M.; Hesse, U. Establishing MinION Sequencing and Genome Assembly Procedures for the Analysis of the Rooibos (Aspalathus linearis) Genome. Plants 2022, 11, 2156. https://doi.org/10.3390/plants11162156
Mgwatyu Y, Cornelissen S, van Heusden P, Stander A, Ranketse M, Hesse U. Establishing MinION Sequencing and Genome Assembly Procedures for the Analysis of the Rooibos (Aspalathus linearis) Genome. Plants. 2022; 11(16):2156. https://doi.org/10.3390/plants11162156
Chicago/Turabian StyleMgwatyu, Yamkela, Stephanie Cornelissen, Peter van Heusden, Allison Stander, Mary Ranketse, and Uljana Hesse. 2022. "Establishing MinION Sequencing and Genome Assembly Procedures for the Analysis of the Rooibos (Aspalathus linearis) Genome" Plants 11, no. 16: 2156. https://doi.org/10.3390/plants11162156
APA StyleMgwatyu, Y., Cornelissen, S., van Heusden, P., Stander, A., Ranketse, M., & Hesse, U. (2022). Establishing MinION Sequencing and Genome Assembly Procedures for the Analysis of the Rooibos (Aspalathus linearis) Genome. Plants, 11(16), 2156. https://doi.org/10.3390/plants11162156