
Genome-Wide Association Study Reveals the Genetic Basis of Kernel and Cob Moisture Changes in Maize at Physiological Maturity Stage

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Descriptions
2.2. SNPs and Candidate Genes Associated with Moisture Changes
2.3. Candidate Gene Association Analysis Revealed Intragenic Variations Affecting Moisture Changes
2.4. Distributions of Superior Alleles in Elite Lines
3. Discussion
3.1. Dissecting the Genetic Basis of Moisture Changes at Physiological Maturity in Maize Using GWAS
3.2. Candidate Genes Involved in MC and DR at Physiological Maturity Stage
3.3. Application of Superior Alleles in Breeding Maize Varieties with Low MC and High DR
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Collection and Data Analysis
4.3. Genome-Wide Association Study
4.4. Candidate Gene Association Study
4.5. Analysis of Superior Alleles
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, G.; Xu, Z.; Wang, F.; Huang, Y.; Xin, Y.; Liang, S.; Li, B.; Si, H.; Sun, L.; Wang, Q.; et al. Development of an efficient and precise adenine base editor (ABE) with expanded target range in allotetraploid cotton (Gossypium hirsutum). BMC Biol. 2022, 20, 45. [Google Scholar] [CrossRef]
- Qian, Y.L.; Zhang, X.Q.; Wang, L.F.; Chen, J.; Chen, B.R.; Lv, G.H.; Wu, Z.C.; Guo, J.; Wang, J.; Qi, Y.C.; et al. Detection of QTLs controlling fast kernel dehydration in maize (Zea mays L.). Genet. Mol. Res. GMR 2016, 15, gmr.15038151. [Google Scholar] [CrossRef]
- Kang, M.S.; Zhang, S. Narrow-Sense Heritability for and Relationship between seed imbibition and grain moisture loss rate in maize. J. New Seeds 2001, 3, 1–16. [Google Scholar] [CrossRef]
- Shaw, R.H.; Loomis, W.E. Bases for the prediction of corn yields. Plant Physiol. 1950, 25, 225–244. [Google Scholar] [CrossRef] [Green Version]
- Capelle, V.; Remoué, C.; Moreau, L.; Reyss, A.; Mahé, A.; Massonneau, A.; Falque, M.; Charcosset, A.; Thévenot, C.; Rogowsky, P.; et al. QTLs and candidate genes for desiccation and abscisic acid content in maize kernels. BMC Plant Biol. 2010, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Ming, B.; Li, L.; Yin, X.; Xue, J.; Wang, K.; Xie, R.; Li, S. Relationship and distribution of in-field dry-down and equilibrium in maize grain moisture content. Agric. For. Meteorol. 2021, 304–305, 108409. [Google Scholar] [CrossRef]
- Xiang, K.; Reid, L.M.; Zhang, Z.-M.; Zhu, X.-Y.; Pan, G.-T. Characterization of correlation between grain moisture and ear rot resistance in maize by QTL meta-analysis. Euphytica 2012, 183, 185–195. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Ding, Y.; Zhang, J.; Zhao, X.; Zhao, F.; Tang, B. Corn kernel dehydration rate: Research progress. J. Agric. 2018, 8, 4–8. [Google Scholar]
- Magari, R.; Kang, M.S.; Zhang, Y. Genotype by environment interaction for ear moisture loss rate in corn. Crop Sci. 1998, 37, 774–779. [Google Scholar]
- Schmidt, J.L.; Hallauer, A.R. Estimating harvest date of corn in the field. Crop Sci. 1966, 6, 227–231. [Google Scholar] [CrossRef]
- Yin, S.; Liu, J.; Yang, T.; Li, P.; Xu, Y.; Fang, H.; Xu, S.; Wei, J.; Xue, L.; Hao, D.; et al. Genetic analysis of the seed dehydration process in maize based on a logistic model. Crop J. 2020, 8, 182–193. [Google Scholar] [CrossRef]
- Liu, J.; Yu, H.; Liu, Y.; Deng, S.; Liu, Q.; Liu, B.; Xu, M. Genetic dissection of grain water content and dehydration rate related to mechanical harvest in maize. BMC Plant Biol. 2020, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.B. Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 2007, 10, 156–161. [Google Scholar] [CrossRef]
- Sukumaran, S.; Dreisigacker, S.; Lopes, M.; Chavez, P.; Reynolds, M.P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 2015, 128, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tan, X.; Yang, Y.; Liu, P.; Zhang, X.; Zhang, Y.; Wang, L.; Hu, Y.; Ma, L.; Li, Z.; et al. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping. Plant Biotechnol. J. 2020, 18, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guan, Z.; Li, Z.; Liu, P.; Ma, L.; Zhang, Y.; Pan, L.; He, S.; Zhang, Y.; Li, P.; et al. A combination of linkage mapping and gwas brings new elements on the genetic basis of yield-related traits in maize across multiple environments. Theor. Appl. Genet. 2020, 133, 2881–2895. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.; Huang, R.; Wu, Y.; Wang, W. The genetic architecture of amylose biosynthesis in maize kernel. Plant Biotechnol. J. 2018, 16, 688–695. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G.; et al. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Ma, L.; An, R.; Jiang, L.; Zhang, C.; Li, Z.; Zou, C.; Yang, C.; Pan, G.; Lübberstedt, T.; Shen, Y. Effects of ZmHIPP on lead tolerance in maize seedlings: Novel ideas for soil bioremediation. J. Hazard. Mater. 2022, 430, 128457. [Google Scholar] [CrossRef]
- Wu, X.; Feng, H.; Wu, D.; Yan, S.; Zhang, P.; Wang, W.; Zhang, J.; Ye, J.; Dai, G.; Fan, Y.; et al. Using high-throughput multiple optical phenotyping to decipher the genetic architecture of maize drought tolerance. Genome Biol. 2021, 22, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, P.; Zhang, X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.; Sun, W.; Guan, Z.; Liang, T.; et al. Multi-locus genome-wide association study reveals the genetic architecture of stalk lodging resistance-related traits in maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.; Gardner, C.; Romay, C.; Ganapathysubramanian, B.; Lübberstedt, T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genom. 2015, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanous, A.; Gardner, C.; Blanco, M.; Martin-Schwarze, A.; Lipka, A.E.; Flint-Garcia, S.; Bohn, M.; Edwards, J.; Lübberstedt, T. Association mapping of flowering and height traits in germplasm enhancement of maize doubled haploid (GEM-DH) lines. Plant Genome 2018, 11, 170083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yu, Y.; Wang, L.; Luo, Y.; Peng, Y.; Xu, Y.; Liu, X.; Wu, S.; Jian, L.; Xu, J.; et al. The genetic architecture of the dynamic changes in grain moisture in maize. Plant Biotechnol. J. 2021, 19, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Y.; Guan, Z.; Liu, P.; He, Y.; Zou, C.; Li, P.; Gao, S.; Peng, H.; Yang, C.; et al. Combined linkage mapping and association analysis reveals genetic control of maize kernel moisture content. Physiol. Plant. 2020, 170, 508–518. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Lu, M.; Yang, D.; Qian, Y.; Yue, Y.; Zhang, Z.; Jin, F.; Wang, M.; Liu, X.; et al. QTL mapping and GWAS for field kernel water content and kernel dehydration rate before physiological maturity in maize. Sci. Rep. 2020, 10, 13114. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Yang, D.; Lu, M.; Qian, Y.; Jin, F.; Liu, X.; Wang, Y.; Liu, W.; Li, X. Detection of QTNs for kernel moisture concentration and kernel dehydration rate before physiological maturity in maize using multi-locus GWAS. Sci. Rep. 2021, 11, 1764. [Google Scholar] [CrossRef]
- Luo, X.; Wang, B.; Gao, S.; Zhang, F.; Terzaghi, W.; Dai, M. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings. J. Integr. Plant Biol. 2019, 61, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Sun, X.; Liu, B.; Kong, F.; Pan, X.; Zhang, H. A polygalacturonase gene PG031 regulates seed coat permeability with a pleiotropic effect on seed weight in soybean. Theor. Appl. Genet. 2022, 135, 1603–1618. [Google Scholar] [CrossRef]
- Klaumann, S.; Nickolaus, S.D.; Fürst, S.H.; Starck, S.; Schneider, S.; Ekkehard Neuhaus, H.; Trentmann, O. The tonoplast copper transporter COPT5 acts as an exporter and is required for interorgan allocation of copper in Arabidopsis Thaliana. New Phytol. 2011, 192, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.T.; Wang, H. Copper toxicity and bioaccumulation in chinese cabbage (Brassica pekinensis Rupr.). Environ. Toxicol. 2005, 20, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Lee, D.G.; Lee, S.H.; Kang, K.Y.; Lee, J.J.; Kim, P.J.; Yoon, H.S.; Kim, J.S.; Lee, B.H. Excess copper induced physiological and proteomic changes in germinating rice seeds. Chemosphere 2007, 67, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.R.; Pichtel, J.; Hayat, S. Copper: Uptake, toxicity and tolerance in plants and management of Cu-contaminated soil. Biometals: Int. J. Role Met. Ions Biol. Biochem. Med. 2021, 34, 737–759. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Liu, H.; Jaqueth, J.; Li, B.; et al. Characterizing the population structure and genetic diversity of maize breeding germplasm in southwest China using genome-wide SNP markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef] [Green Version]
- Dou, X. Advances in studies on the function of plant polygalacturonase. J. Shanxi Agric. Univ. Nat. Sci. Ed. 2004, 24, 5. [Google Scholar]
- Nie, H.; Shi, Y.; Geng, X.; Xing, G. CRISRP/Cas9-mediated targeted mutagenesis of tomato polygalacturonase gene (SlPG) delays fruit softening. Front. Plant Sci. 2022, 13, 729128. [Google Scholar] [CrossRef]
- Zhang, G.; Hou, X.; Wang, L.; Xu, J.; Chen, J.; Fu, X.; Shen, N.; Nian, J.; Jiang, Z.; Hu, J.; et al. Photo-sensitive leaf rolling 1 encodes a polygalacturonase that modifies cell wall structure and drought tolerance in rice. New Phytol. 2021, 229, 890–901. [Google Scholar] [CrossRef]
- Liang, T.; Qing, C.; Liu, P.; Zou, C.; Yuan, G.; Pan, G.; Shen, Y.; Ma, L. Joint GWAS and WGCNA uncover the genetic control of calcium accumulation under salt treatment in maize seedlings. Physiol. Plant. 2022, 174, e13606. [Google Scholar] [CrossRef]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C.; et al. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Kaler, A.S.; Gillman, J.D.; Beissinger, T.; Purcell, L.C. Comparing different statistical models and multiple testing corrections for association mapping in soybean and maize. Front. Plant Sci. 2019, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Becker, L.C.; Becker, D.M.; Starmer, J.D.; Province, M.A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet. Epidemiol. 2010, 34, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.C.; Nelson, G.W.; Troyer, J.L.; Lautenberger, J.A.; Kessing, B.D.; Winkler, C.A.; O’Brien, S.J. Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genom. 2010, 11, 724. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef]
- Zhang, X.; Guan, Z.; Wang, L.; Fu, J.; Zhang, Y.; Li, Z.; Ma, L.; Liu, P.; Zhang, Y.; Liu, M.; et al. Combined GWAS and QTL analysis for dissecting the genetic architecture of kernel test weight in maize. Mol. Genet. Genom. MGG 2020, 295, 409–420. [Google Scholar] [CrossRef]

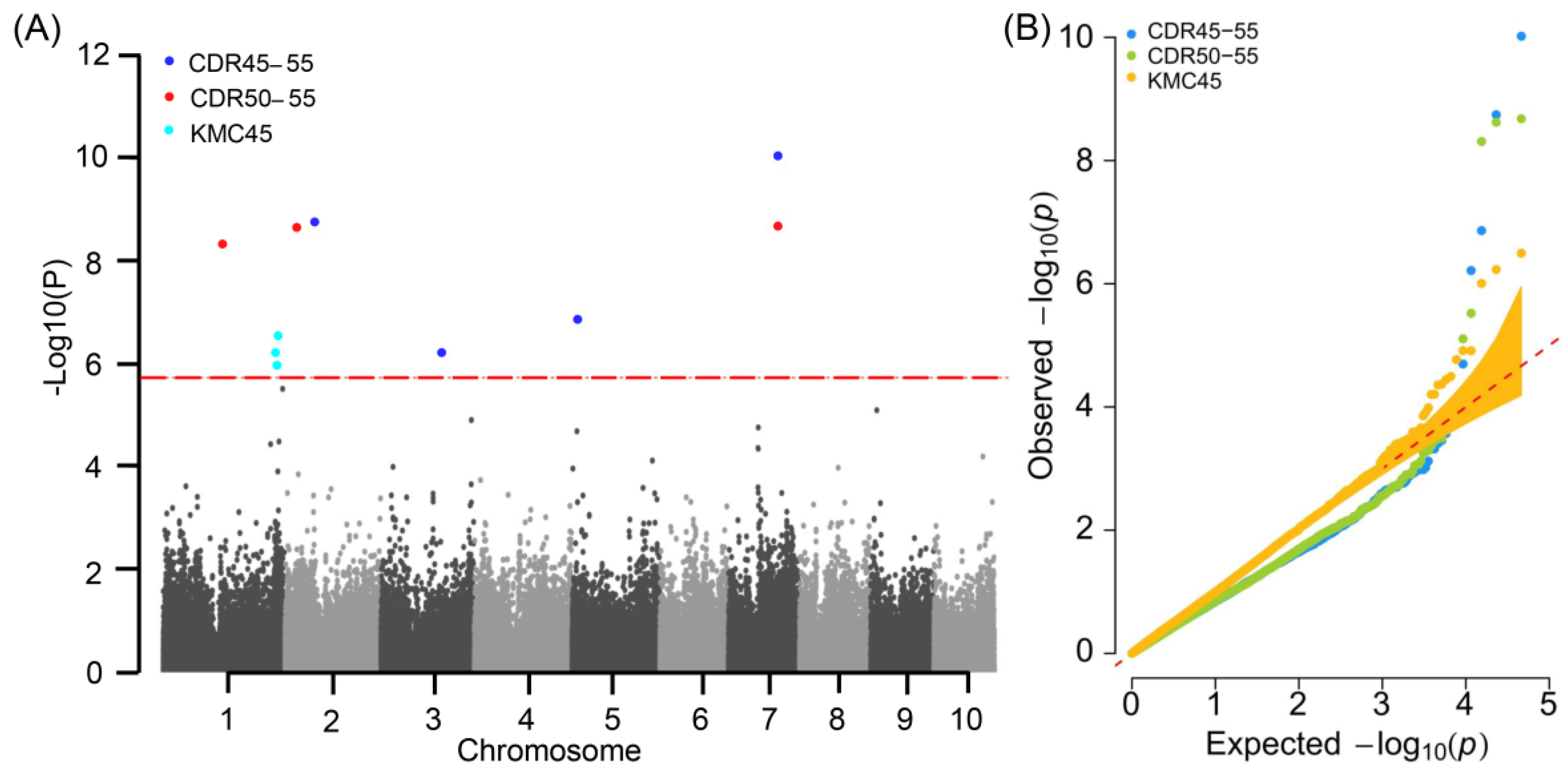

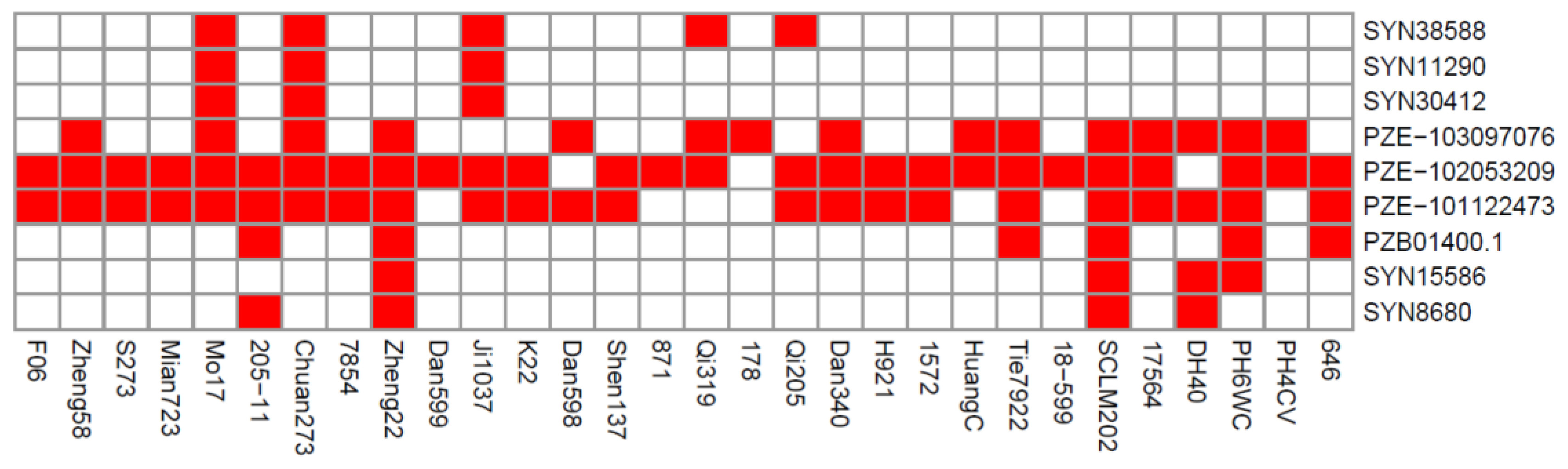

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean (%) | Max (%) | Min (%) | SD | CV (%) | H2 (%) |
|---|---|---|---|---|---|---|
| CMC45 | 62.60 | 78.75 | 46.37 | 0.38 | 0.61 | 62.38 |
| CMC50 | 61.26 | 77.28 | 42.30 | 0.48 | 0.79 | 73.69 |
| CMC55 | 57.01 | 71.80 | 35.52 | 0.71 | 1.25 | 65.25 |
| KMC45 | 35.70 | 73.50 | 22.52 | 0.49 | 1.37 | 29.79 |
| KMC50 | 32.59 | 65.55 | 14.46 | 0.49 | 1.50 | 35.15 |
| KMC55 | 26.98 | 46.72 | 11.46 | 0.61 | 2.26 | 68.71 |
| CDR45–50 | 0.95 | 4.78 | 0.01 | 0.08 | 8.15 | 92.16 |
| CDR50–55 | 1.11 | 4.53 | 0.02 | 0.10 | 9.35 | 88.41 |
| CDR45–55 | 0.72 | 3.41 | 0.01 | 0.08 | 10.68 | 60.41 |
| KDR45–50 | 0.85 | 1.84 | 0.06 | 0.04 | 7.51 | 22.20 |
| KDR50–55 | 1.16 | 2.56 | 0.05 | 0.08 | 8.75 | 24.52 |
| KDR45–55 | 0.75 | 1.36 | 0.06 | 0.04 | 6.33 | 65.41 |
| Associated Traits | Candidate Genes | Annotations |
|---|---|---|
| Zm00001d020618 | - | |
| Zm00001d020610 | - | |
| Zm00001d020609 | - | |
| Zm00001d020612 | - | |
| Zm00001d020622 | - | |
| Zm00001d020615 | polygalacturonase | |
| CDR45–55 | Zm00001d020626 | - |
| CDR50–55 | Zm00001d020613 | - |
| Zm00001d020617 | exocyst complex component EXO70B1 | |
| Zm00001d020614 | - | |
| Zm00001d020623 | copper transporter 5.1 | |
| Zm00001d020616 | - | |
| Zm00001d020627 | - | |
| Zm00001d020628 | trimethyltridecatetraene synthase | |
| Zm00001d020620 | - |
| Trait | Marker | Position | p-Value | Allele | Variation Region |
|---|---|---|---|---|---|
| CDR50–55 | S7_125165191 | 125165191 | 0.00259 | G/A | the third exon (synonymous) |
| KDR45–50 | S7_125166053 | 125166053 | 0.00246 | C/T | the fifth exon (synonymous) |
| S7_125166495 | 125166495 | 0.00108 | C/A | the fifth exon (missense) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Xiangchen, C.; Yan, J.; Chengxu, Y.; Liu, H.; Zou, C.; Pan, G.; Shen, Y.; Ma, L. Genome-Wide Association Study Reveals the Genetic Basis of Kernel and Cob Moisture Changes in Maize at Physiological Maturity Stage. Plants 2022, 11, 1989. https://doi.org/10.3390/plants11151989
Zhang M, Xiangchen C, Yan J, Chengxu Y, Liu H, Zou C, Pan G, Shen Y, Ma L. Genome-Wide Association Study Reveals the Genetic Basis of Kernel and Cob Moisture Changes in Maize at Physiological Maturity Stage. Plants. 2022; 11(15):1989. https://doi.org/10.3390/plants11151989
Chicago/Turabian StyleZhang, Minyan, Chaoyang Xiangchen, Jiaquan Yan, Yujuan Chengxu, Hao Liu, Chaoying Zou, Guangtang Pan, Yaou Shen, and Langlang Ma. 2022. "Genome-Wide Association Study Reveals the Genetic Basis of Kernel and Cob Moisture Changes in Maize at Physiological Maturity Stage" Plants 11, no. 15: 1989. https://doi.org/10.3390/plants11151989
APA StyleZhang, M., Xiangchen, C., Yan, J., Chengxu, Y., Liu, H., Zou, C., Pan, G., Shen, Y., & Ma, L. (2022). Genome-Wide Association Study Reveals the Genetic Basis of Kernel and Cob Moisture Changes in Maize at Physiological Maturity Stage. Plants, 11(15), 1989. https://doi.org/10.3390/plants11151989