
All Set before Flowering: A 16S Gene Amplicon-Based Analysis of the Root Microbiome Recruited by Common Bean (Phaseolus vulgaris) in Its Centre of Domestication




Abstract
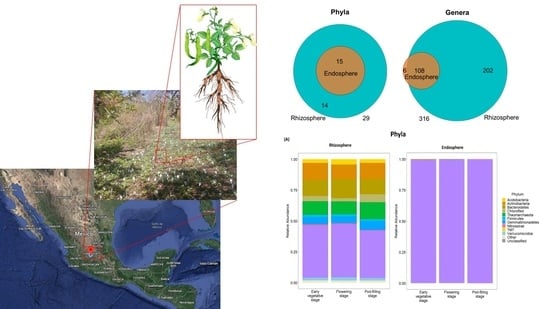
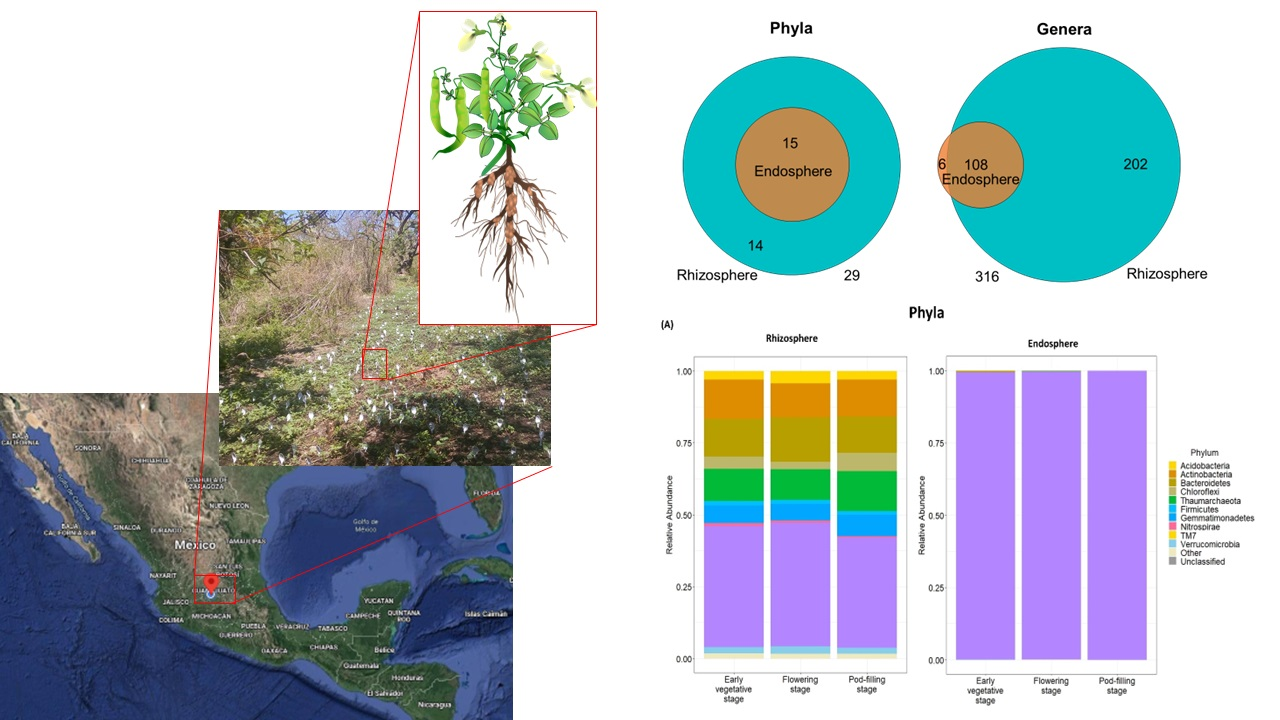
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Compartment | Conditions | Plant Stage | 16S * | Size | Main Result | Ref |
|---|---|---|---|---|---|---|---|
| 2010 | Endosphere (seed and roots) | Sterile | Seedling 3 d | primers fD1 rD1 | 1500 bp | Dominating phyla: Firmicutes, Actinobacteria, α-β-and γ-Proteobacteria; genera: Acinetobacter, Bacillus, Methylobacterium, Micrococcus, Paenibacillus, Rhizobium, Staphylococcus | [53] |
| 2017 | Rhizosphere | Pots, GH 8 genotypes (2 WA, 3 LR, 3 MC) | Flowering | V3–V4 | 460 bp | Genotype explained only 13% of bact. diversity, MC harboured less Bacteroidetes than wild accessions and landraces | [59] |
| 2018 | Rhizosphere | Pots, GH, Amazon dark earth + agric. soil, 2 cultivars diff resistance to F. oxy. | Flowering | V3–V4 & shotgun metagenome | 460 bp | More diverse and connected community in the F. oxy-resistant cultivar, dominated by Pseudomonadaceae, Bacillaceae, Solibacteraceae and Cytophagaceae. | [60] |
| 2019 | Rhizosphere | Pots, GH, Amazon dark earth + agric. soil 8 genotypes (2 WA, 3 LR, 3 MC) | Flowering | V3–V4 | 460 bp | Effects of bean genotype stronger in the agricultural soil, among a total of 15,925 OTUs, 113 highly abundant OTUs (26% of all reads) conform a core microbiome shared by all accession x soil combinations | [24] |
| 2020 | Endosphere and Rhizosphere | Open field ex-situ | Flowering | primers fD1 rD1 | 1500 bp | 12 out of 90 cultured strains exhibited direct antibiosis: 7 Bacillus, 2 Pseudomonas, 1 Agrobacterium, 1 Glutamicibacter | [52] |
| 2020 | Endosphere and bulk soil | Pots, GH 2 genotypes w. different root morphology | Plantlet 15 d | V3 | No difference between genotypes, OTU richness and diversity in soil much higher than in vermiculite | [61] | |
| 2021 | Rhizosphere and bulk soil | Open field ex-situ, 2 cultivars | Vegetative Flowering Pod filling Pods ripe | V4–V5 | 300 bp | Weak/no effect of plant genotype, while location and soil properties as main determinants generate a biogeographic pattern of bacterial community structures | [37] |
| 2022 | Rhizosphere and bulk soil | Open field ex-situ, 2 cultivars | Vegetative | V4 | 290 bp | More cultivar-exclusive than shared OTUs, taxa rhizosphere of biofortified cultivar enriched in diverse groups, e.g., Burkholderia and Rhodanobacter | [62] |
| 2022 | Seed endosphere | 1 cultivar | Seeds | V4 | 300 bp | Bacterial seed endosphere communities show inter- but not intra-individual variation | [63] |
| 2022 | Endosphere and Rhizosphere | Open field in domestication area | Vegetative Flowering Pod filling | V5–V9 | 750 bp | This study |
2. Results and Discussion
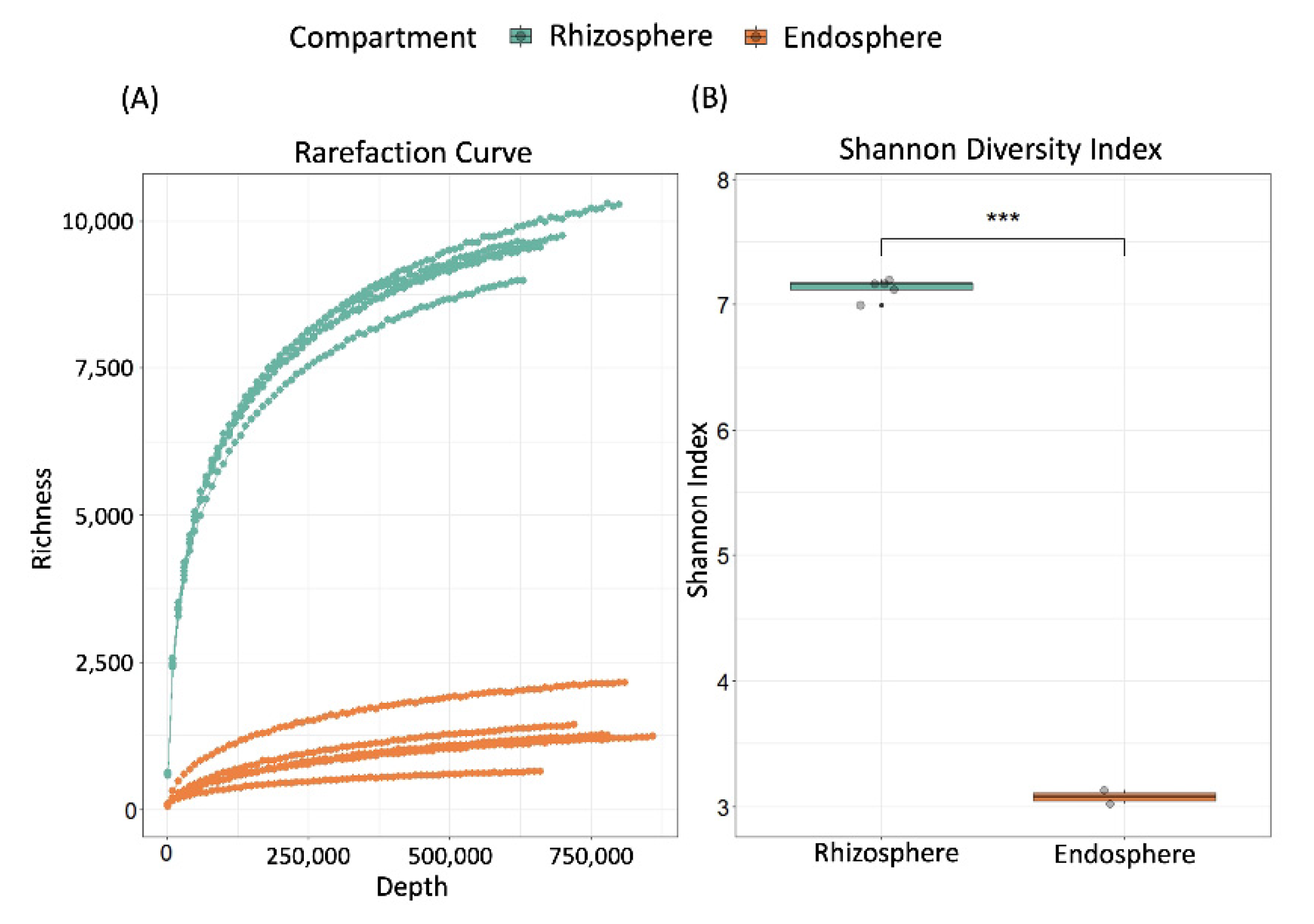
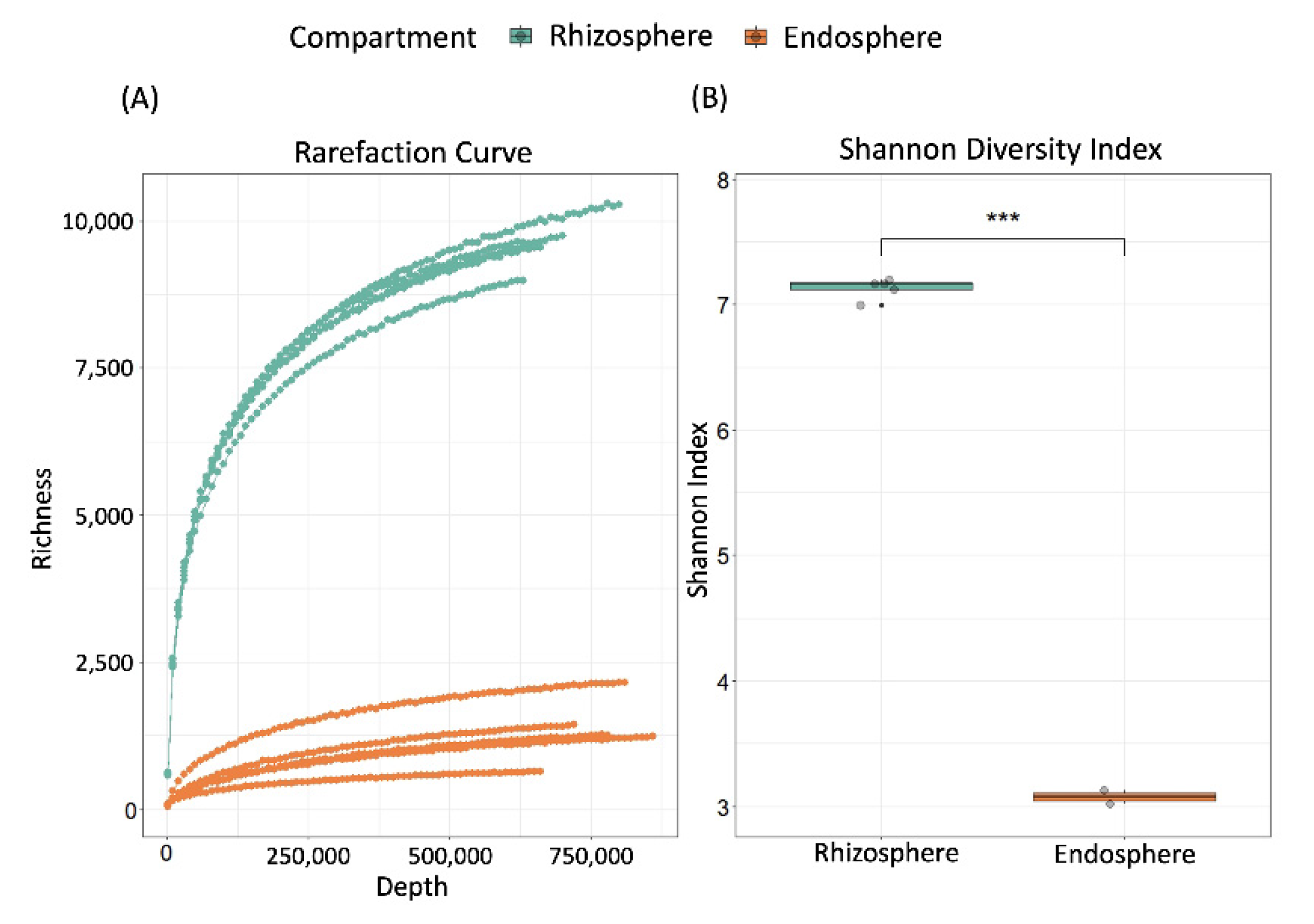
2.1. Validation of Experimental Design and Overall Bacterial Diversity
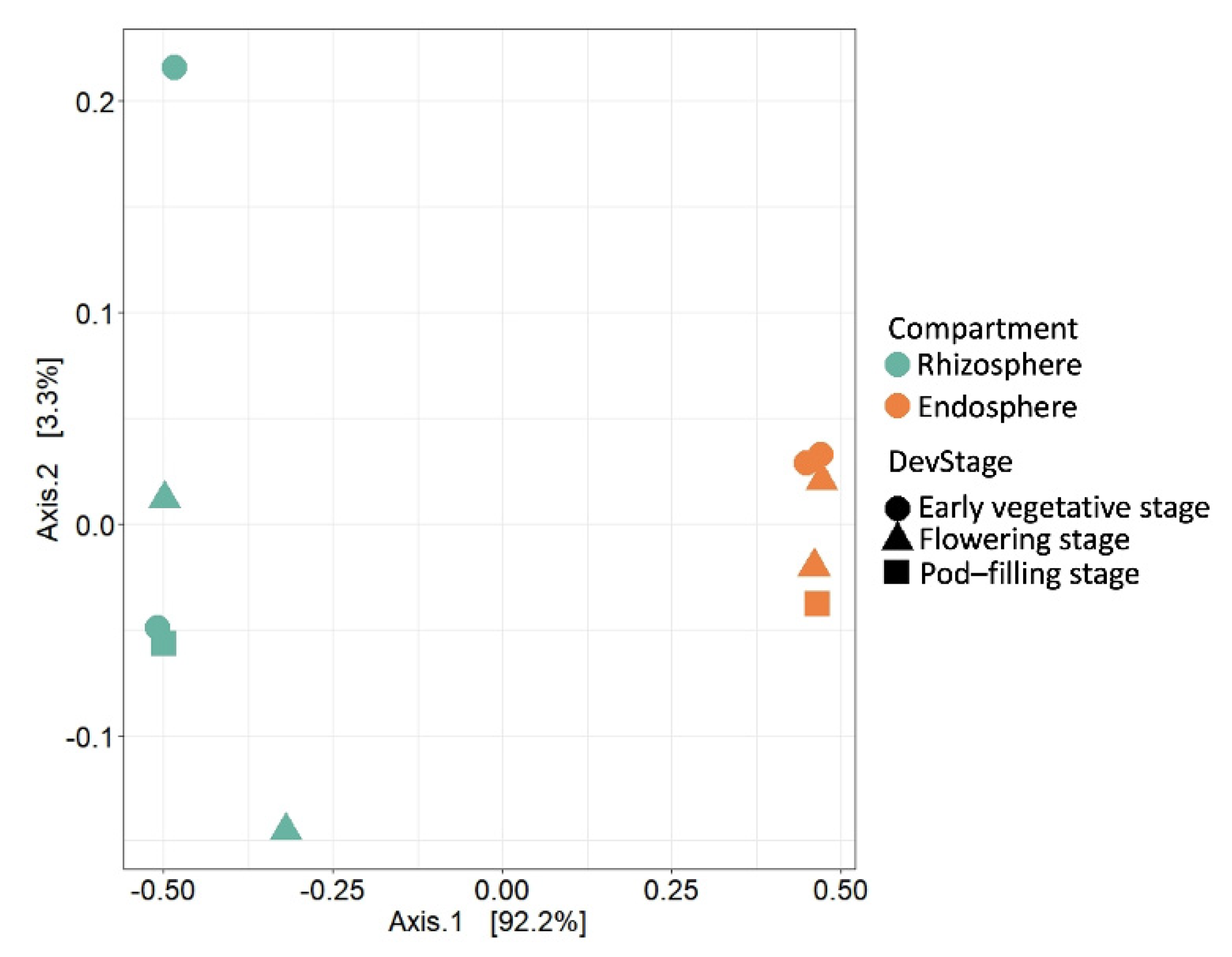
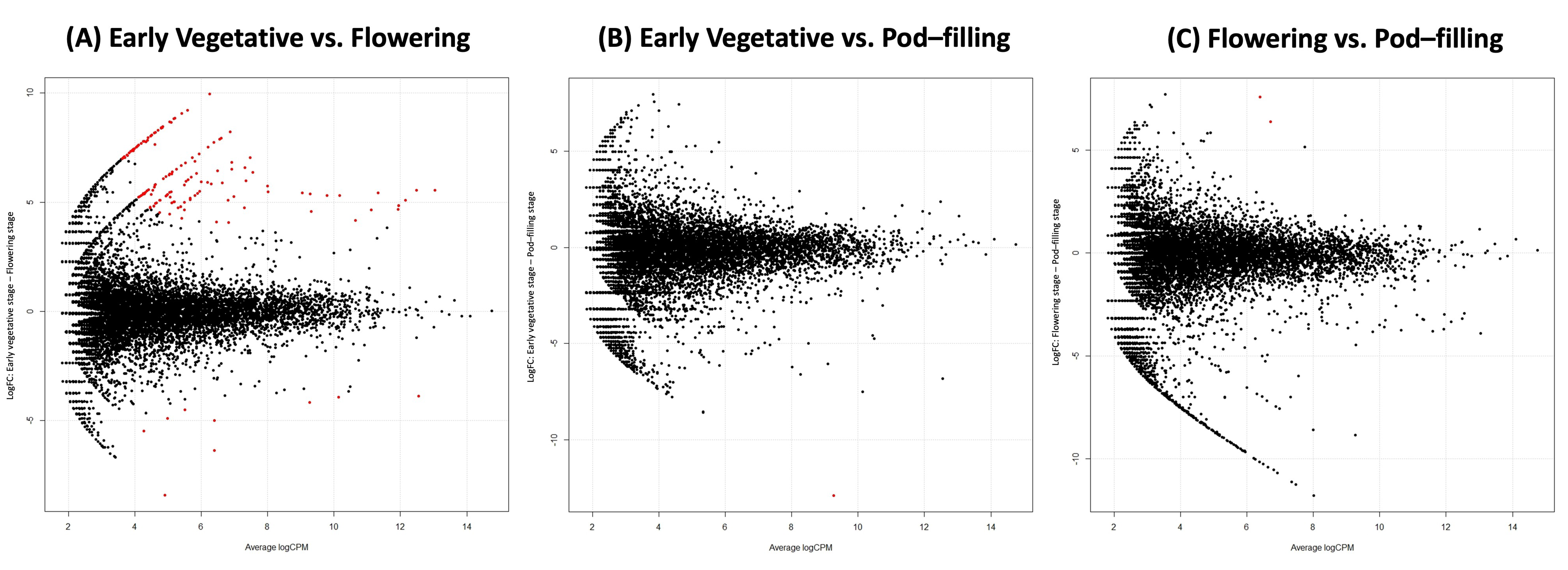
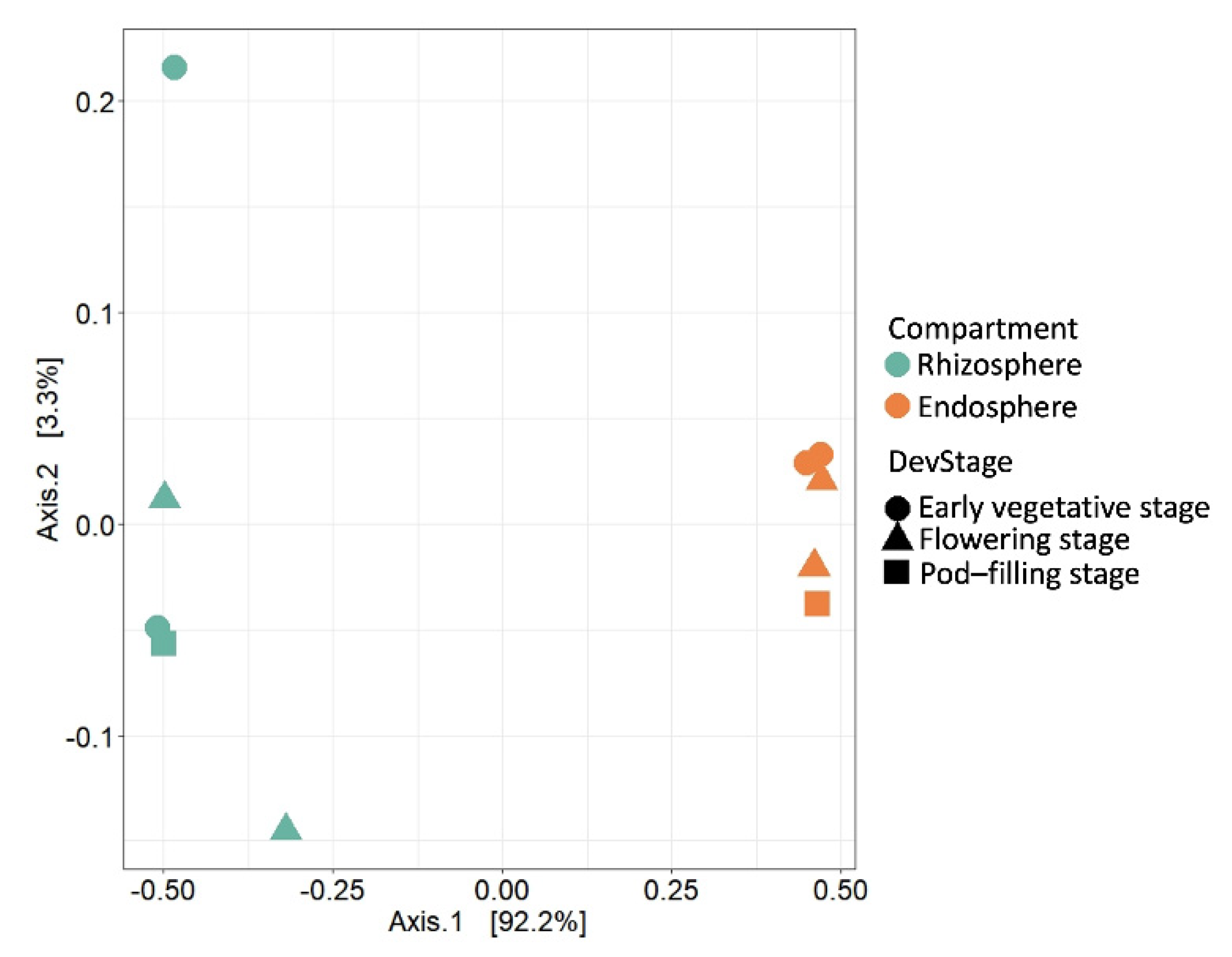
2.2. Compartment Rather Than Ontogenetic Stage Determines the Composition of Bean Prokaryotic Microbiota
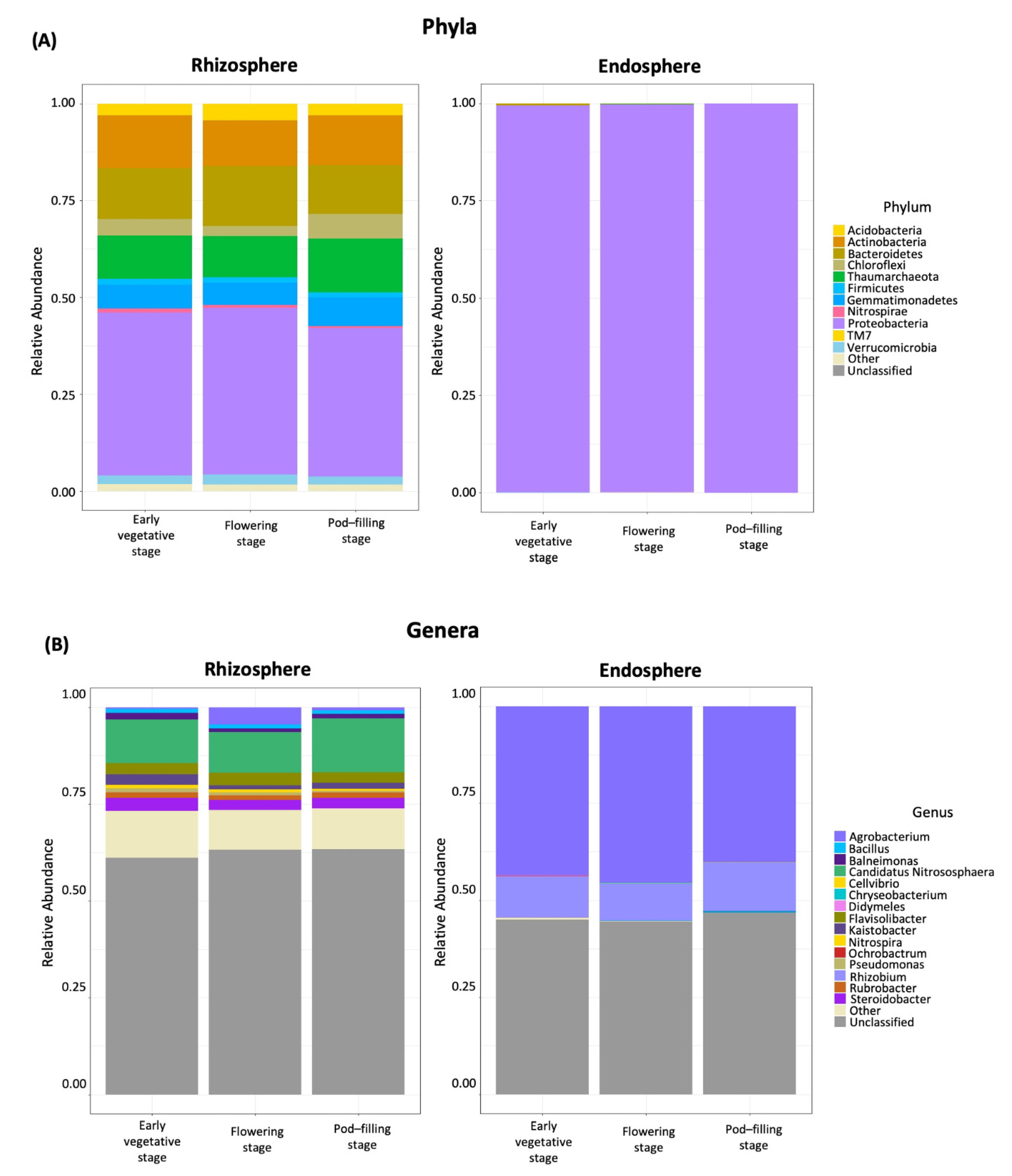
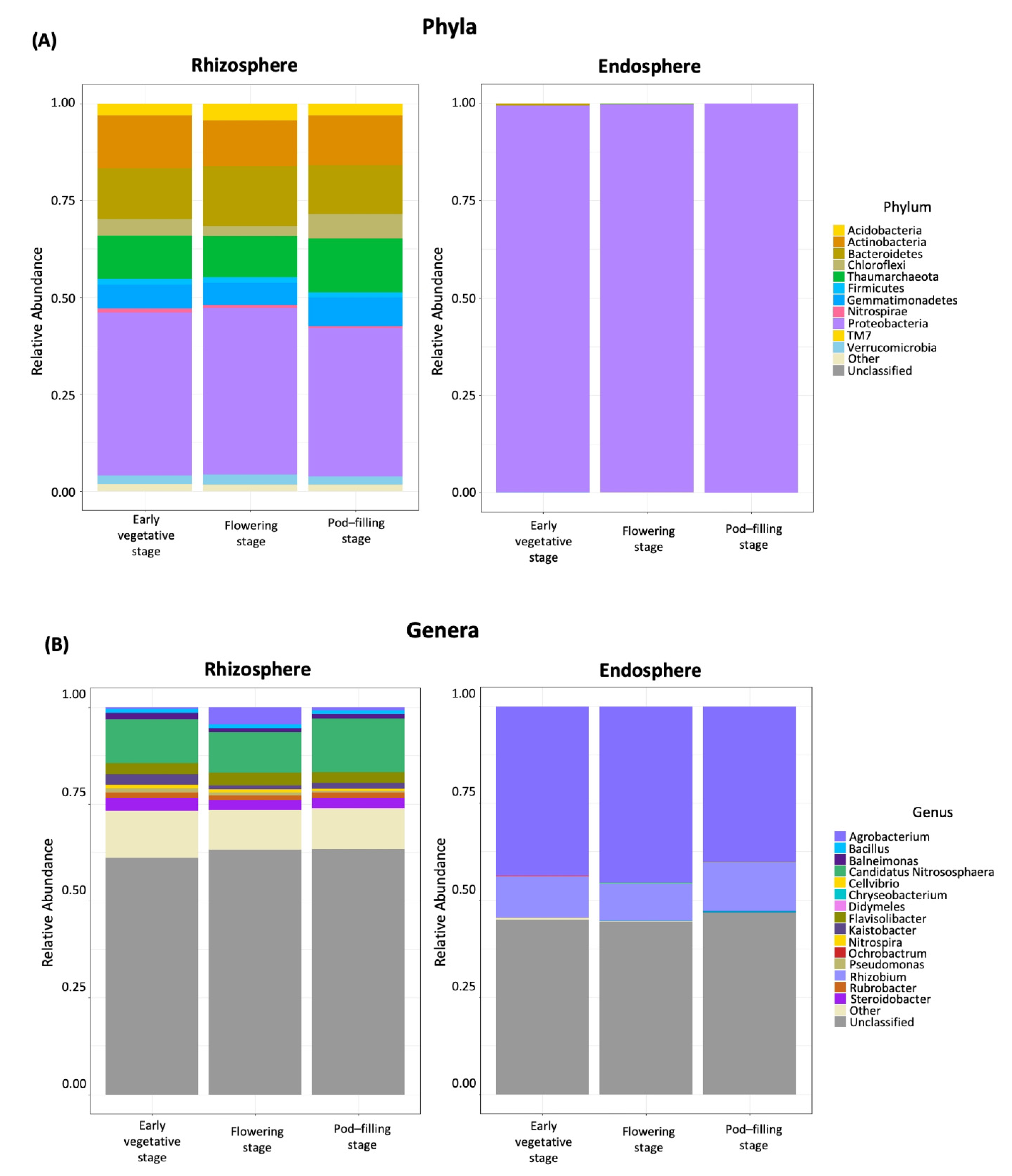
2.3. Dominant Taxa in Rhizosphere versus Endosphere
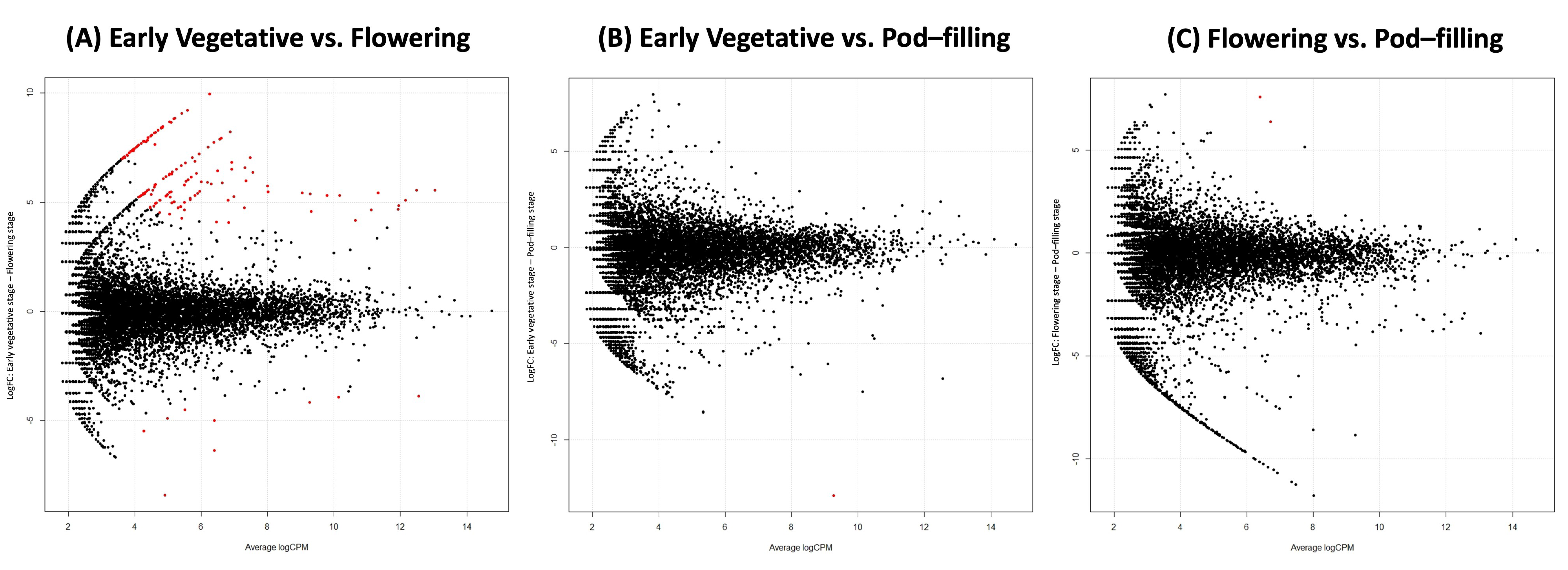
2.4. Ontogenetic Shift from Stenotrophomonas to N-Fixing Taxa in the Rhizosphere
| ID | Family | Genus | Read Numbers | |
|---|---|---|---|---|
| Vegetative Stage | Flowering Stage | |||
| 1083508 | Xanthomonadaceae | Stenotrophomonas | 18640 | 1298 |
| 537062 | Xanthomonadaceae | Stenotrophomonas | 3507 | 238 |
| 227343 | Xanthomonadaceae | Stenotrophomonas | 1926 | 110 |
| 4045633 | Rhizobiaceae | Rhizobium | 555 | 27149 |
| 843074 | Rhizobiaceae | Rhizobium | 465 | 12328 |
| 1104546 | Rhizobiaceae | Rhizobium | 401 | 12232 |
| 155854 | Rhizobiaceae | Rhizobium | 400 | 14421 |
| 714181 | Rhizobiaceae | Rhizobium | 365 | 18057 |
| 225582 | Rhizobiaceae | Rhizobium | 267 | 4970 |
| 1107243 | Rhizobiaceae | Rhizobium | 263 | 6907 |
| 80113 | Rhizobiaceae | Rhizobium | 179 | 8120 |
| 591708 | Xanthomonadaceae | Stenotrophomonas | 92 | 3 |
| 848768 | Rhizobiaceae | Rhizobium | 87 | 3650 |
| 220539 | Rhizobiaceae | Rhizobium | 79 | 1976 |
| 634321 | Flavobacteriaceae | Flavobacterium | 67 | 2 |
| 200464 | Rhizobiaceae | Rhizobium | 46 | 2024 |
| 1104627 | Rhizobiaceae | Rhizobium | 37 | 1685 |
| 370368 | Rhodobacteraceae | Paracoccus | 10 | 435 |
| 529216 | Rhodobacteraceae | Paracoccus | 4 | 103 |
| 833408 | Aeromonadaceae | Aeromonas | 3 | 203 |
| 38159 | Rhizobiaceae | Rhizobium | 3 | 78 |
| 834097 | Aeromonadaceae | Aeromonas | 2 | 129 |
| 1085832 | Streptococcaceae | Streptococcus | 2 | 94 |
| 813705 | Aeromonadaceae | Aeromonas | 2 | 88 |
| 831599 | Aeromonadaceae | Aeromonas | 1 | 151 |
| 837574 | Aeromonadaceae | Aeromonas | 1 | 60 |
| 423025 | Aeromonadaceae | Aeromonas | 1 | 54 |
| 564995 | Rhizobiaceae | Rhizobium | 1 | 51 |
| 1141678 | Aeromonadaceae | Aeromonas | 1 | 48 |
| 578911 | Bacillaceae | Exiguobacterium | 0 | 108 |
| 641892 | Blastocatellaceae | Aridibacter | 0 | 102 |
| 171996 | Rhodobacteraceae | Paracoccus | 0 | 89 |
| 830659 | Streptococcaceae | Lactococcus | 0 | 62 |
| 165293 | Cyclobacteriaceae | Algoriphagus | 0 | 58 |
| 388951 | Aeromonadaceae | Aeromonas | 0 | 55 |
| 1144093 | Hyphomicrobiales | Liberibacter | 0 | 41 |
| 1143479 | Cyclobacteriaceae | Algoriphagus | 0 | 38 |
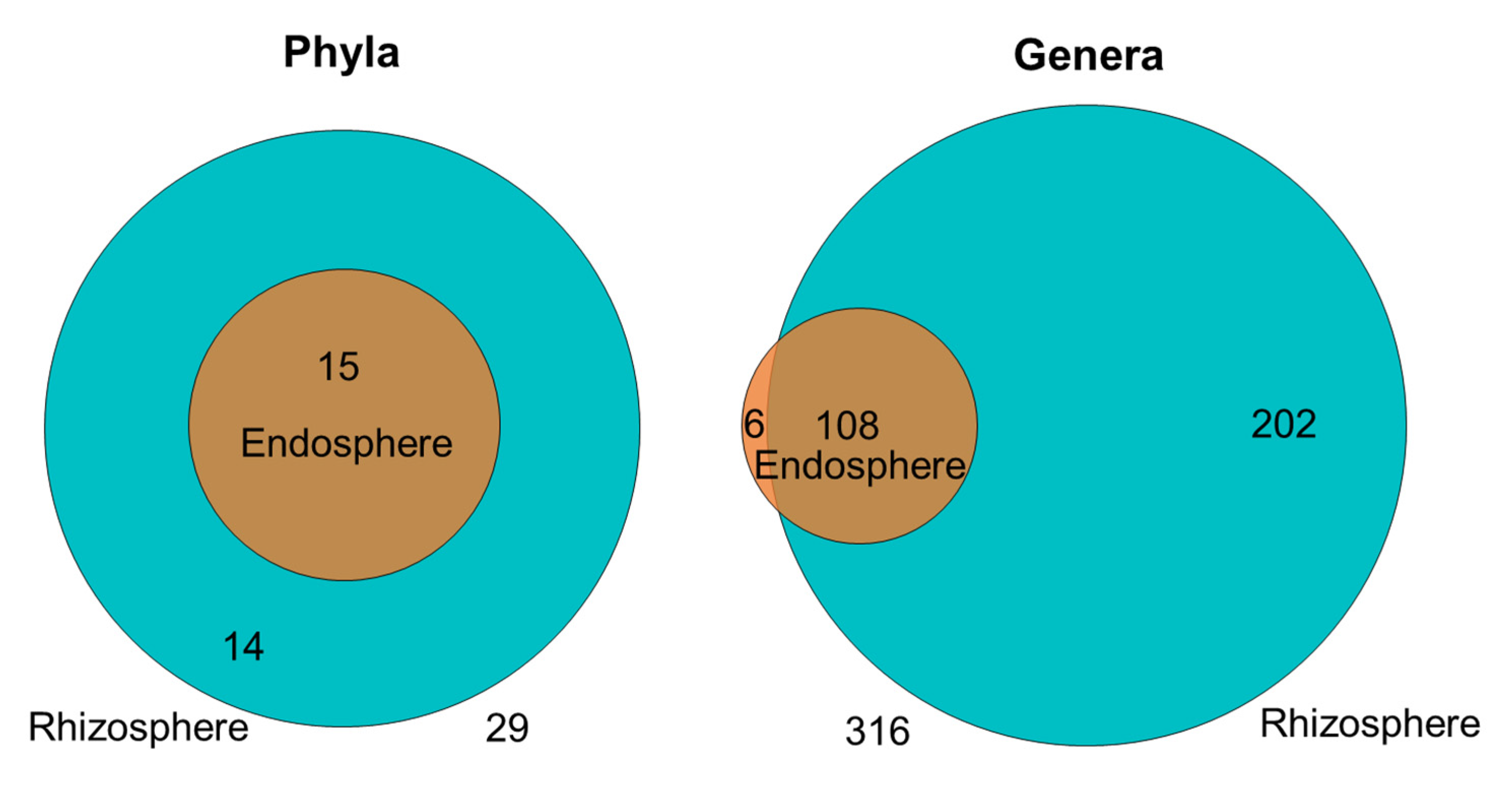
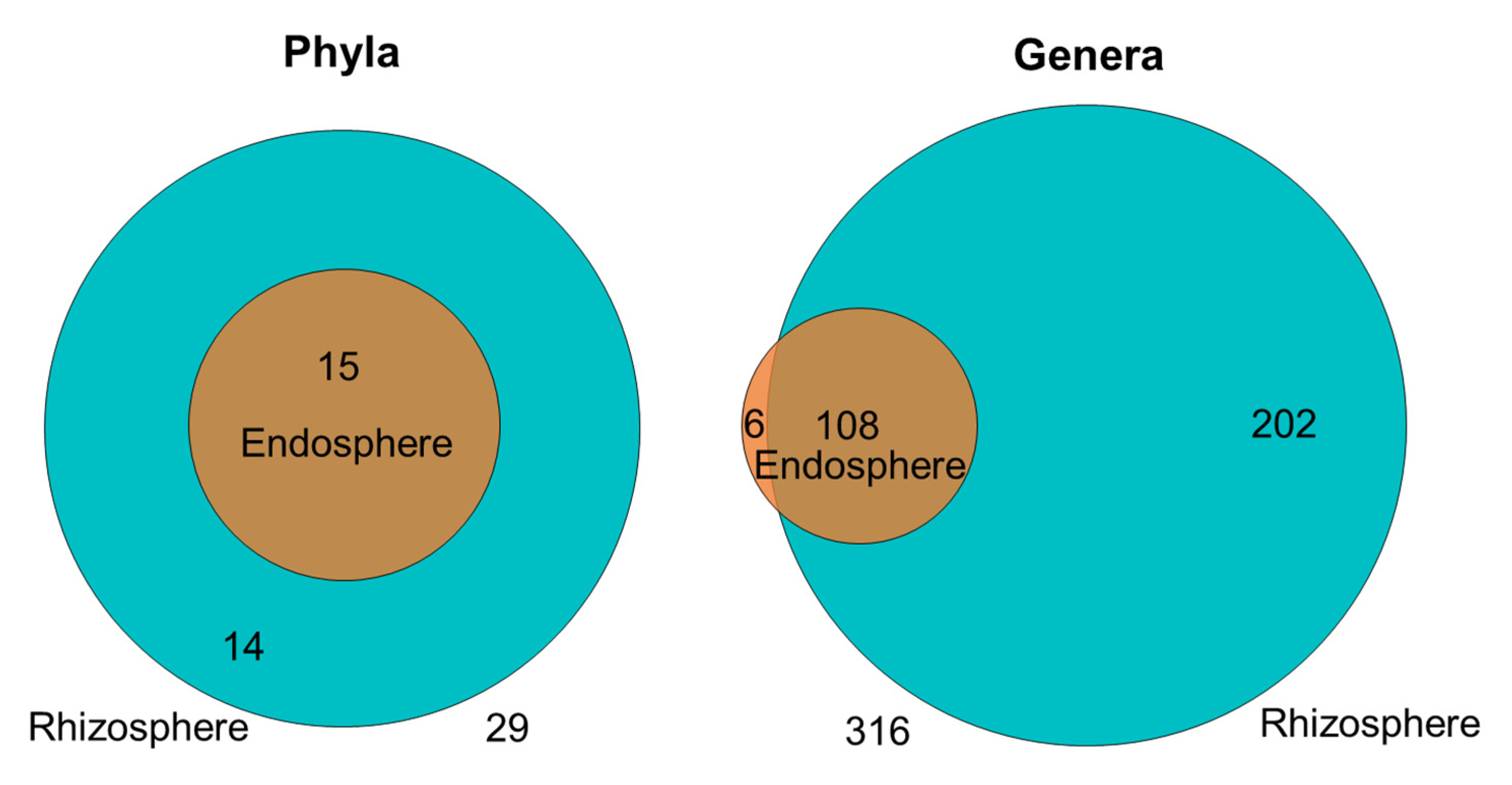
2.5. Factors Explaining the Differentiation between Endosphere and Rhizosphere Bacterial Communities Assembled from the Native Inoculum
2.5.1. The Bean Root Endosphere Is Colonized by a Subset of the Rhizosphere Community
2.5.2. Asking “What Can They Do?” and “What Must They Do?” to Identify the Controlling Partner
2.5.3. Things Bacteria MUST Do for Themselves and Things They CAN Do for the Plant
2.5.4. Things Bacteria MUST Decide Who Dominates the Bean Rhizosphere
| Functional Group/KO | Gene | KEGG ID | Sign. | Enrichment | Comment | Ref | |
|---|---|---|---|---|---|---|---|
| O | E | ||||||
| Beneficial for bacterium | |||||||
| Degradation of plant polymers | |||||||
| Endoglucanase | - | K01179 | *** | Required to penetrate root and cell surfaces and—eventually—for the liberation of nutrients from abundant plant structural molecules | [5,92,106] | ||
| Endo-1,3(4)-β-glucanase | - | K01180 | * | ||||
| Endo-1,4-β-xylanase | xynA | K01181 | * | ||||
| Oligo-1,6-glucosidase | malL | K01182 | *** | ||||
| Polygalacturonase | - | K01184 | ** | ||||
| Xylan 1,4-β-xylosidase | xynB | K01198 | *** | ||||
| Licheninase | bglS | K01216 | *** | ||||
| Stress-related enzymes | |||||||
| Glutathione peroxidase | btuE | K00432 | *** | Detoxification of ROS | [106] | ||
| Glutathione S-transferase | gst | K00799 | *** | ||||
| Catalase | katE | K03781 | ** | ||||
| Secretion systems | |||||||
| Type II | gspD | K02453 | *** | Injection of effectors into eukaryotic host cells to suppress host immunity | [106,112,113] | ||
| Type IV | virB2 | K03197 | *** | ||||
| Type VI | hcp | K05601 | *** | ||||
| Type I pilus assembly | fimA | K07345 | ** | ||||
| Chemotaxis and motility | |||||||
| Chemotaxis protein | motA | K02556 | ** | General chemotaxis | |||
| Serine | tsr | K05874 | ** | Carbon source chemotaxis | [92] | ||
| Aspartate/maltose | tar | K05875 | *** | ||||
| Ribose | rbsB | K10439 | *** | ||||
| Galactose | mglB | K10540 | *** | ||||
| Flagellar apparatus | fliI | K02412 | (*) | Overrepresented in rice endosphere | [5,106] | ||
| Twitching motility | chpA | K06596 | *** | Usually considered relevant to colonize the endosphere | [6,114] | ||
| pilJ | K02660 | *** | |||||
| Signal trans. 2-comp systems | |||||||
| Carbon source utilization | creC | K07641 | ** | Carbon source utilization and toxin resistance required for rhizosphere colonization | |||
| creB | K07663 | *** | |||||
| Multidrug resistance | baeS | K07642 | *** | [4,6] | |||
| baeR | K07664 | *** | |||||
| Antibiotic resistance | evgS | K07679 | *** | ||||
| evgA | K07690 | *** | |||||
| Amino sugar metabolism | glrK | K07711 | *** | ||||
| Cell fate control | pleD | K02488 | *** | [115] | |||
| pleC | K07716 | *** | |||||
| Beneficial for both | |||||||
| Plant growth promotion | |||||||
| Acetoin reductase | budC | K03366 | *** | Synthesis of 2,3-butanediol, VOC, growth and resistance induction, bacterial survival | [116,117,118] | ||
| Acetolactate decarboxylase | alsD | K01575 | *** | ||||
| Butanediol dehydrogenase | butB | K00004 | *** | ||||
| Plant hormones | |||||||
| S-adenosylmethionine syt. | metK | K00789 | *** | Ethylene for suppression of plant resistance, indole acetic acid (IAA) enhances bacterial rhizosphere competence | [5,92] | ||
| IAA biosyn. IAM pathway | amiE | K01426 | *** | [119,120] | |||
| IAA biosyn. IPyA pathway | ipdC | K04103 | *** | ||||
| Degradation of microbial polymers | |||||||
| Chitinase | - | K01183 | *** | Antibiosis, microbe-microbe competition, or ‘biocontrol’ of microbial pathogens | [4,6] | ||
| Chitinase/lysozyme | chiA | K13381 | *** | ||||
| Service for plant | |||||||
| nod genes and nitrogen fixation | |||||||
| LysR family TF | nodD | K14657 | *** | Nod genes required for plant colonization and nodule formation, nifA controls the nif operon, nitrogenase nifH, for nitrogen fixation, 2 component system proteins ntrY and ntrX control nitrogen fixation and metabolism | [92] | ||
| Nodulation protein | nodA | K14658 | *** | ||||
| Chitooligos-deacetyl | nodB | K14659 | *** | ||||
| nodulation protein | nodE | K14660 | *** | ||||
| nodF | K14661 | *** | |||||
| GlcNAc transferase | nodC | K14666 | *** | ||||
| Nif regulatory protein | nifA | K02584 | *** | ||||
| Nitrogenase | nifH | K02588 | ns | ||||
| Histidine kinase | ntrY | K13598 | *** | ||||
| N regul. response factor | ntrX | K13599 | *** | ||||
| Nutrient solubilizing | |||||||
| 3-phytase | - | K01083 | * | ||||
3. Materials and Methods
3.1. Plant Material, Growing Conditions, and Sampling
3.2. DNA Extraction from Soil (Rhizosphere) and Root (Endosphere) Samples
3.3. Generation of 16S rRNA Gene Amplicons
3.4. Bioinformatic Data Processing and OTU Picking
3.5. Rarefaction Curves and Diversity Analysis
3.6. Statistical Analysis and Differential Abundances
3.7. Prediction of Functional Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hacquard, S.; Garrido-Oter, R.; González, A.; Spaepen, S.; Ackermann, G.; Lebeis, S.; McHardy, A.C.; Dangl, J.L.; Knight, R.; Ley, R.; et al. Microbiota and Host Nutrition across Plant and Animal Kingdoms. Cell Host Microbe 2015, 17, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Kuzyakov, Y.; Razavi, B.S. Rhizosphere size and shape: Temporal dynamics and spatial stationarity. Soil Biol. Biochem. 2019, 135, 343–360. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots Shaping Their Microbiome: Global Hotspots for Microbial Activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Carvalhais, L.C.; Crawford, M.; Singh, E.; Dennis, P.G.; Pieterse, C.M.J.; Schenk, P.M. Inner plant values: Diversity, colonization and benefits from endophytic bacteria. Front. Microbiol. 2017, 8, 2552. [Google Scholar] [CrossRef]
- Afzal, I.; Shinwari, Z.K.; Sikandar, S.; Shahzad, S. Plant beneficial endophytic bacteria: Mechanisms, diversity, host range and genetic determinants. Microbiol. Res. 2019, 221, 36–49. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The Hidden World within Plants: Ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [Green Version]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Vieira, S.; Sikorski, J.; Dietz, S.; Herz, K.; Schrumpf, M.; Bruelheide, H.; Scheel, D.; Friedrich, M.W.; Overmann, J. Drivers of the composition of active rhizosphere bacterial communities in temperate grasslands. ISME J. 2020, 14, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Tkacz, A.; Bestion, E.; Bo, Z.; Hortala, M.; Poole, P.S. Influence of Plant Fraction, Soil, and Plant Species on Microbiota: A Multikingdom Comparison. mBio 2020, 11, e02785-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Hulbert, S.H.; Schroeder, K.L.; Mavrodi, O.; Mavrodi, D.; Dhingra, A.; Schillinger, W.F.; Paulitz, T.C. Role of Bacterial Communities in the Natural Suppression of Rhizoctonia solani Bare Patch Disease of Wheat (Triticum aestivum L.). Appl. Environ. Microbiol. 2013, 79, 7428–7438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barea, J.-M.; Pozo, M.J.; Azcon, C.; Azcón-Aguilar, C. Microbial co-operation in the rhizosphere. J. Exp. Bot. 2005, 56, 1761–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, R.; Ulanova, D.; Wick, L.Y.; Bode, H.B.; Garbeva, P. Microbe-driven chemical ecology: Past, present and future. ISME J. 2019, 13, 2656–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busby, P.E.; Soman, C.; Wagner, M.; Friesen, M.; Kremer, J.; Bennett, A.; Morsy, M.; Eisen, J.A.; Leach, J.E.; Dangl, J.L. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 2017, 15, e2001793. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Hinojosa, A.; Strauss, S.L. Insights into the taxonomic and functional characterization of agricultural crop core rhizobiomes and their potential microbial drivers. Sci. Rep. 2021, 11, 10068. [Google Scholar] [CrossRef]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Ezachow, C.; Müller, H.; Etilcher, R.; Berg, G. Differences between the rhizosphere microbiome of Beta vulgaris ssp. maritima—Ancestor of all beet crops—And modern sugar beets. Front. Microbiol. 2014, 5, 415. [Google Scholar] [CrossRef] [Green Version]
- Leff, J.W.; Lynch, R.C.; Kane, N.C.; Fierer, N. Plant domestication and the assembly of bacterial and fungal communities associated with strains of the common sunflower, Helianthus annuus. New Phytol. 2016, 214, 412–423. [Google Scholar] [CrossRef]
- Brisson, V.L.; Schmidt, J.E.; Northen, T.R.; Vogel, J.P.; Gaudin, A.C.M. Impacts of Maize Domestication and Breeding on Rhizosphere Microbial Community Recruitment from a Nutrient Depleted Agricultural Soil. Sci. Rep. 2019, 9, 15611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston-Monje, D.; Raizada, M.N. Conservation and Diversity of Seed Associated Endophytes in Zea across Boundaries of Evolution, Ethnography and Ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Jaramillo, J.E.; De Hollander, M.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M.; Carrión, V.J. Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 2019, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.A.; Jin, Z.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. USA 2018, 115, 7368–7373. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.; Zilber-Rosenberg, I. Microbes Drive Evolution of Animals and Plants: The Hologenome Concept. mBio 2016, 7, e01395-15. [Google Scholar] [CrossRef] [Green Version]
- Hale, I.L.; Broders, K.; Iriarte, G. A Vavilovian approach to discovering crop-associated microbes with potential to enhance plant immunity. Front. Plant Sci. 2014, 5, 492. [Google Scholar] [CrossRef] [Green Version]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca-Garcia, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida-Martinez, L.P.; Tringe, S.G. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 2015, 209, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Rybakova, D.; Mancinelli, R.; Wikström, M.; Birch-Jensen, A.-S.; Postma, J.; Ehlers, R.-U.; Goertz, S.; Berg, G. The structure of the Brassica napus seed microbiome is cultivar-dependent and affects the interactions of symbionts and pathogens. Microbiome 2017, 5, 104. [Google Scholar] [CrossRef]
- Hernández-Álvarez, C.; García-Oliva, F.; Cruz-Ortega, R.; Romero, M.F.; Barajas, H.R.; Piñero, D.; Alcaraz, L.D. Squash root microbiome transplants and metagenomic inspection for in situ arid adaptations. Sci. Total Environ. 2021, 805, 150136. [Google Scholar] [CrossRef] [PubMed]
- Coats, V.C.; Pelletreau, K.N.; Rumpho, M.E. Amplicon pyrosequencing reveals the soil microbial diversity associated with invasive Japanese barberry (Berberis thunbergii DC.). Mol. Ecol. 2013, 23, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Aires, T.; Stuij, T.M.; Muyzer, G.; Serrão, E.A.; Engelen, A.H. Characterization and Comparison of Bacterial Communities of an Invasive and Two Native Caribbean Seagrass Species Sheds Light on the Possible Influence of the Microbiome on Invasive Mechanisms. Front. Microbiol. 2021, 12, 2123. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.S.; Snoek, L.B.; Koorem, K.; Geisen, S.; Bloem, L.J.; ten Hooven, F.; Kostenko, O.; Krigas, N.; Manrubia, M.; Caković, D.; et al. Range-expansion effects on the belowground plant microbiome. Nat. Ecol. Evol. 2019, 3, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhu, F.; Carrión, V.J.; Cordovez, V. Beyond Plant Microbiome Composition: Exploiting Microbial Functions and Plant Traits via Integrated Approaches. Front. Bioeng. Biotechnol. 2020, 8, 896. [Google Scholar] [CrossRef] [PubMed]
- Coats, V.C.; Rumpho, M.E. The rhizosphere microbiota of plant invaders: An overview of recent advances in the microbiomics of invasive plants. Front. Microbiol. 2014, 5, 368. [Google Scholar] [CrossRef] [PubMed]
- Stopnisek, N.; Shade, A. Persistent microbiome members in the common bean rhizosphere: An integrated analysis of space, time, and plant genotype. ISME J. 2021, 15, 2708–2722. [Google Scholar] [CrossRef]
- Wagner, M.R. Prioritizing host phenotype to understand microbiome heritability in plants. New Phytol. 2021, 232, 502–509. [Google Scholar] [CrossRef]
- Agler, M.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation. PLOS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [Green Version]
- Dardanelli, M.S.; de Córdoba, F.J.F.; Estévez, J.; Contreras, R.; Cubo, M.T.; Rodriguez-Carvajal, M.A.; Gil-Serrano, A.M.; Baena, F.J.L.; Bellogín, R.; Manyani, H.; et al. Changes in flavonoids secreted by Phaseolus vulgaris roots in the presence of salt and the plant growth-promoting rhizobacterium Chryseobacterium balustinum. Appl. Soil Ecol. 2012, 57, 31–38. [Google Scholar] [CrossRef]
- Han, Q.; Ma, Q.; Chen, Y.; Tian, B.; Xu, L.; Bai, Y.; Chen, W.; Li, X. Variation in rhizosphere microbial communities and its association with the symbiotic efficiency of rhizobia in soybean. ISME J. 2020, 14, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.L.; Mejia, D. Phaseolus Bean: Post-Harvest Operations in INPhO Post Harvest Compendium; Centro Internacional de Agricultura Tropical (CIAT), Ed.; Food and Agriculture Organisation (FAO) of the United Nations: Rome, Italy, 1999; p. 24. [Google Scholar]
- Brown, C.H.; Clement, C.; Epps, P.; Luedeling, E.; Wichmann, S. The Paleobiolinguistics of the Common Bean (Phaseolus vulgaris L.). Ethnobiol. Lett. 2014, 5, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Gepts, P.; Bliss, F.A. F1 hybrid weakness in the common bean: Differential geographic origin suggets two gene pools in cultivated bean germplasm. J. Hered. 1985, 76, 447–450. [Google Scholar] [CrossRef]
- Rendón-Anaya, M.; Montero-Vargas, J.M.; Saburido-Álvarez, S.; Vlasova, A.; Capella-Gutierrez, S.; Ordaz-Ortiz, J.J.; Aguilar, O.M.; Vianello-Brondani, R.P.; Santalla, M.; Delaye, L.; et al. Genomic history of the origin and domestication of common bean unveils its closest sister species. Genome Biol. 2017, 18, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitocchi, E.; Rau, D.; Bellucci, E.; Rodriguez, M.; Murgia, M.L.; Gioia, T.; Santo, D.; Nanni, L.; Attene, G.; Papa, R. Beans (Phaseolus ssp.) as a Model for Understanding Crop Evolution. Front. Plant Sci. 2017, 8, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.; Kami, J.A.; Gepts, P. The Putative Mesoamerican Domestication Center of Phaseolus vulgaris Is Located in the Lerma–Santiago Basin of Mexico. Crop Sci. 2009, 49, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Debouck, D.G.; Toro, O.; Paredes, O.M.; Johnson, W.C.; Gepts, P. Genetic diversity and ecological distribution of Phaseolus vulgaris (Fabaceae) in northwestern South America. Econ. Bot. 1993, 47, 408–423. [Google Scholar] [CrossRef]
- Peralta, H.; Aguilar, A.; Díaz, R.; Mora, Y.; Martínez-Batallar, G.; Salazar, E.; Vargas-Lagunas, C.; Martínez, E.; Encarnación, S.; Girard, L.; et al. Genomic studies of nitrogen-fixing rhizobial strains from Phaseolus vulgaris seeds and nodules. BMC Genom. 2016, 17, 711. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Romero, E. Coevolution in Rhizobium-Legume Symbiosis? DNA Cell Biol. 2009, 28, 361–370. [Google Scholar] [CrossRef]
- Castro-Guerrero, N.A.; Isidra-Arellano, M.C.; Mendoza-Cozatl, D.G.; Valdés-López, O. Common Bean: A Legume Model on the Rise for Unraveling Responses and Adaptations to Iron, Zinc, and Phosphate Deficiencies. Front. Plant Sci. 2016, 7, 600. [Google Scholar] [CrossRef] [Green Version]
- Sendi, Y.; Pfeiffer, T.; Koch, E.; Mhadhbi, H.; Mrabet, M. Potential of common bean (Phaseolus vulgaris L.) root microbiome in the biocontrol of root rot disease and traits of performance. J. Plant Dis. Prot. 2020, 127, 453–462. [Google Scholar] [CrossRef]
- López-López, A.; Rogel, M.A.; Ormeño-Orrillo, E.; Martinez-Romero, J.C.; Martínez-Romero, E. Phaseolus vulgaris seed-borne endophytic community with novel bacterial species such as Rhizobium endophyticum sp. nov. Syst. Appl. Microbiol. 2010, 33, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Alström, S. Induction of disease resistance in common bean susceptible to halo blight bacterial pathogen after seed bacterization with rhizosphere Pseudomonas. J. Gen. Appl. Microbiol. 1991, 37, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; De Queiroz, M.V.; Borges, A.C.; De Moraes, C.A.; De Araújo, E.F. Isolation and characterization of endophytic bacteria isolated from the leaves of the common bean (Phaseolus vulgaris). Braz. J. Microbiol. 2012, 43, 1562–1575. [Google Scholar] [CrossRef]
- Martins, S.J.; De Medeiros, F.H.V.; De Souza, R.M.; De Resende, M.L.V.; Ribeiro, P.M. Biological control of bacterial wilt of common bean by plant growth-promoting rhizobacteria. Biol. Control 2013, 66, 65–71. [Google Scholar] [CrossRef]
- Martins, S.A.; Schurt, D.A.; Seabra, S.S.; Martins, S.J.; Ramalho, M.A.P.; Moreira, F.; Silva, J.C.P.; da Silva, J.A.G.; Medeiros, F. Common bean (Phaseolus vulgaris L.) growth promotion and biocontrol by rhizobacteria under Rhizoctonia solani suppressive and conducive soils. Appl. Soil Ecol. 2018, 127, 129–135. [Google Scholar] [CrossRef]
- Negi, S.; Bharat, N.K.; Kaushal, R.; Rohiwala, P. Screening of bioagents for seed biopriming in French bean (Phaseolus vulgaris L.) under Laboratory condition. Int. J. Chem. Stud. 2020, 8, 790–793. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Carrion, V.J.; Bosse, M.; Ferrão, L.F.V.; De Hollander, M.; Garcia, A.A.F.; Ramirez, C.A.; Mendes, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef] [Green Version]
- Mendes, L.W.; Raaijmakers, J.M.; De Hollander, M.; Mendes, R.; Tsai, S.M. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2018, 12, 212–224. [Google Scholar] [CrossRef]
- Barraza, A.; Vizuet-De-Rueda, J.C.; Alvarez-Venegas, R. Highly diverse root endophyte bacterial community is driven by growth substrate and is plant genotype-independent in common bean (Phaseolus vulgaris L.). PeerJ 2020, 8, e9423. [Google Scholar] [CrossRef]
- Cardoso, A.M.; da Silva, C.V.F.; Albano, R.M.; Pádua, V.L.M. Bacterial communities in the rhizosphere of biofortified BRS pontal and conventional carioca bean (Phaseolus vulgaris) plants. Arch. Microbiol. 2021, 204, 14. [Google Scholar] [CrossRef] [PubMed]
- Bintarti, A.F.; Sulesky-Grieb, A.; Stopnišek, N.; Shade, A. Endophytic Microbiome Variation Among Single Plant Seeds. Phytobiomes J. 2022, 6, 45–55. [Google Scholar] [CrossRef]
- Blair, M.W.; Díaz, L.M.; Gill-Langarica, H.R.; Rosales-Serna, R.; Mayek-Perez, N.; Acosta-Gallegos, J.A. Genetic Relatedness of Mexican Common Bean Cultivars Revealed by Microsatellite Markers. Crop Sci. 2011, 51, 2655–2667. [Google Scholar] [CrossRef]
- Gottel, N.R.; Castro, H.F.; Kerley, M.; Yang, Z.; Pelletier, D.; Podar, M.; Karpinets, T.; Uberbacher, E.; Tuskan, G.A.; Vilgalys, R.; et al. Distinct Microbial Communities within the Endosphere and Rhizosphere of Populus deltoides Roots across Contrasting Soil Types. Appl. Environ. Microbiol. 2011, 77, 5934–5944. [Google Scholar] [CrossRef] [Green Version]
- Beckers, B.; Op De Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s rDNA Primer Pairs in the Study of Rhizosphere and Endosphere Bacterial Microbiomes in Metabarcoding Studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Chelius, M.K.; Triplett, E.W. The Diversity of Archaea and Bacteria in Association with the Roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef]
- Dos Santos, H.R.M.; Argolo, C.S.; Argôlo-Filho, R.C.; Loguercio, L.L. A 16S rDNA PCR-based theoretical to actual delta approach on culturable mock communities revealed severe losses of diversity information. BMC Microbiol. 2019, 19, 74. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Elvia, J.C.; de Freitas, R.J.; Germida, J.J. Bacterial Microbiomes Associated with the Rhizosphere, Root Interior, and Aboveground Plant Organs of Wheat and Canola at Different Growth Stages. Phytobiomes J. 2021, 5, 442–451. [Google Scholar] [CrossRef]
- Yamazaki, S.; Mardani-Korrani, H.; Kaida, R.; Ochiai, K.; Kobayashi, M.; Nagano, A.J.; Fujii, Y.; Sugiyama, A.; Aoki, Y. Field multi-omics analysis reveals a close association between bacterial communities and mineral properties in the soybean rhizosphere. Sci. Rep. 2021, 11, 8878. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.-H.; Sang, M.K.; Song, J.; Weon, H.-Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Chen, W.; Zong, L.; Yang, J.; Jiao, S.; Lin, Y.; Wang, E.T.; Wei, G. Two cultivated legume plants reveal the enrichment process of the microbiome in the rhizocompartments. Mol. Ecol. 2017, 26, 1641–1651. [Google Scholar] [CrossRef] [PubMed]
- Hartman, K.; Van Der Heijden, M.G.; Roussely-Provent, V.; Walser, J.-C.; Schlaeppi, K. Deciphering composition and function of the root microbiome of a legume plant. Microbiome 2017, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, L.; Singh, R.P.; Meng, C.; Ma, S.; Jing, C.; Li, Y.; Zhang, C. Nodule and Root Zone Microbiota of Salt-Tolerant Wild Soybean in Coastal Sand and Saline-Alkali Soil. Front. Microbiol. 2020, 11, 2178. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Shao, T.; Long, X.; He, T.; Gao, X.; Zhou, Z.; Liu, Z.; Rengel, Z. Microbiome structure and function in rhizosphere of Jerusalem artichoke grown in saline land. Sci. Total Environ. 2020, 724, 138259. [Google Scholar] [CrossRef] [PubMed]
- Gkarmiri, K.; Mahmood, S.; Ekblad, A.; Alström, S.; Högberg, N.; Finlay, R. Identifying the Active Microbiome Associated with Roots and Rhizosphere Soil of Oilseed Rape. Appl. Environ. Microbiol. 2017, 83, e01938-17. [Google Scholar] [CrossRef] [Green Version]
- Cordero, J.; De Freitas, J.R.; Germida, J. Bacterial microbiome associated with the rhizosphere and root interior of crops in Saskatchewan, Canada. Can. J. Microbiol. 2020, 66, 71–85. [Google Scholar] [CrossRef]
- Li, X.; Garvey, M.; Kaplan, I.; Li, B.; Carrillo, J. Domestication of tomato has reduced the attraction of herbivore natural enemies to pest-damaged plants. Agric. For. Èntomol. 2018, 20, 390–401. [Google Scholar] [CrossRef]
- Pester, M.; Schleper, C.; Wagner, M. The Thaumarchaeota: An emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 2011, 14, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.T.; Berg-Lyons, D.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2010, 5, 908–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.J.; Michel, F.C.; Hadar, Y.; Minz, D. Contrasting patterns of seed and root colonization by bacteria from the genus Chryseobacterium and from the family Oxalobacteraceae. ISME J. 2007, 1, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathma, J.; Sakthivel, N. Molecular and functional characterization of bacteria isolated from straw and goat manure based vermicompost. Appl. Soil Ecol. 2013, 70, 33–47. [Google Scholar] [CrossRef]
- Gardener, B.B.M.; Weller, D.M. Changes in Populations of Rhizosphere Bacteria Associated with Take-All Disease of Wheat. Appl. Environ. Microbiol. 2001, 67, 4414–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattenburger, C.J.; Halverson, L.J.; Hofmockel, K.S. Agricultural Management Affects Root-Associated Microbiome Recruitment over Maize Development. Phytobiomes J. 2019, 3, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.A.; Santos-Medellin, C.M.; Liechty, Z.S.; Nguyen, B.; Lurie, E.; Eason, S.; Phillips, G.; Sundaresan, V. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018, 16, e2003862. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.-Y.; Rahman, A.; Li, H.-X.; Xu, T.; Ding, G.-C.; Li, J. Modification of total and phosphorus mineralizing bacterial communities associated with Zea mays L. through plant development and fertilization regimes. J. Integr. Agric. 2021, 20, 3026–3038. [Google Scholar] [CrossRef]
- Oberhaensli, T.; Hofer, V.; Tamm, L.; Fuchs, J.; Koller, M.; Herforth-Rahmé, J.; Maurhofer, M.; Thuerig, B. Aeromonas media in compost amendments contributes to suppression of Pythium ultimum in cress. Acta Hortic. 2017, 1164, 353–360. [Google Scholar] [CrossRef]
- Inbar, J.; Chet, I. Evidence that chitinase produced by Aeromonas caviae is involved in the biological control of soil-borne plant pathogens by this bacterium. Soil Biol. Biochem. 1991, 23, 973–978. [Google Scholar] [CrossRef]
- Brígido, C.; Singh, S.; Menéndez, E.; Tavares, M.J.; Glick, B.R.; Félix, M.D.R.; Oliveira, S.; Carvalho, M. Diversity and Functionality of Culturable Endophytic Bacterial Communities in Chickpea Plants. Plants 2019, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Rosenblueth, M.; Martinez-Romero, E. Bacterial Endophytes and Their Interactions with Hosts. Mol. Plant-Microbe Interact. 2006, 19, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi, P.; Starcher, M.R.; Thallinger, G.G.; Zachow, C.; Müller, H.; Berg, G. Stenotrophomonas comparative genomics reveals genes and functions that differentiate beneficial and pathogenic bacteria. BMC Genom. 2014, 15, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gao, J.; Bai, Z.; Wu, S.; Li, X.; Wang, N.; Du, X.; Fan, H.; Zhuang, G.; Bohu, T.; et al. Unraveling Mechanisms and Impact of Microbial Recruitment on Oilseed Rape (Brassica napus L.) and the Rhizosphere Mediated by Plant Growth-Promoting Rhizobacteria. Microorganisms 2021, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Suckstorff, I.; Berg, G. Evidence for dose-dependent effects on plant growth by Stenotrophomonas strains from different origins. J. Appl. Microbiol. 2003, 95, 656–663. [Google Scholar] [CrossRef] [Green Version]
- Ajilogba, C.F.; Olanrewaju, O.S.; Babalola, O.O. Plant Growth Stage Drives the Temporal and Spatial Dynamics of the Bacterial Microbiome in the Rhizosphere of Vigna subterranea. Front. Microbiol. 2022, 13, 825377. [Google Scholar] [CrossRef]
- Berg, G.; Raaijmakers, J. Saving seed microbiomes. ISME J. 2018, 12, 1167–1170. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Reis, F.; Richards, C.L.; Bossdorf, O. Understanding plant microbiomes requires a genotype × environment framework. Am. J. Bot. 2021, 108, 1820–1823. [Google Scholar] [CrossRef]
- Levy, A.; Salas Gonzalez, I.; Mittelviefhaus, M.; Clingenpeel, S.; Harrera Paredes, S.; Miao, J.; Wang, K.; Devescovi, G.; Stillman, K.; Monteiro, F.; et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 2018, 50, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Pieterse, C.M.; de Jonge, R.; Berendsen, R. The Soil-Borne Supremacy. Trends Plant Sci. 2016, 21, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jaramillo, J.E.; Carrion, V.J.; De Hollander, M.; Raaijmakers, J.M. The wild side of plant microbiomes. Microbiome 2018, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, O.M.; Castrillo, G.; Paredes, S.H.; González, I.S.; Dangl, J.L. Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 2017, 38, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Ucros, J.; Zwetsloot, M.J.; Cuellar-Gempeler, C.; Bauerle, T.L. Spatiotemporal patterns of rhizosphere microbiome assembly: From ecological theory to agricultural application. J. Appl. Ecol. 2021, 58, 894–904. [Google Scholar] [CrossRef]
- Sessitsch, A.; Hardoim, P.; Doering, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M.; et al. Functional Characteristics of an Endophyte Community Colonizing Rice Roots as Revealed by Metagenomic Analysis. Mol. Plant-Microbe Interact. 2012, 25, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Partida-Martinez, L.P.P.; Heil, M. The Microbe-Free Plant: Fact or Artifact? Front. Plant Sci. 2011, 2, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbeva, P.; Weisskopf, L. Airborne medicine: Bacterial volatiles and their influence on plant health. New Phytol. 2019, 226, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Langille, M.G.I. A primer and discussion on DNA-based microbiome data and related bioinformatics analyses. Peer Community J. 2021, 1, 4. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Martiny, A.C.; Treseder, K.; Pusch, G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013, 7, 830–838. [Google Scholar] [CrossRef]
- Song, G.C.; Jeon, J.; Choi, H.K.; Sim, H.; Kim, S.; Ryu, C. Bacterial type III effector–induced plant C8 volatiles elicit antibacterial immunity in heterospecific neighbouring plants via airborne signalling. Plant Cell Environ. 2021, 45, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S.; Spaepen, S.; Garrido-Oter, R.; Schulze-Lefert, P. Interplay Between Innate Immunity and the Plant Microbiota. Annu. Rev. Phytopathol. 2017, 55, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.J.; Feltcher, M.E.; Waters, R.J.; Wetmore, K.M.; Mucyn, T.S.; Ryan, E.M.; Wang, G.; Ul-Hasan, S.; McDonald, M.; Yoshikuni, Y.; et al. Genome-wide identification of bacterial plant colonization genes. PLoS Biol. 2017, 15, e2002860. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Schäper, S.; Becker, A. Cyclic di-GMP signaling controlling the free-living lifestyle of alpha-proteobacterial rhizobia. Biol. Chem. 2020, 401, 1335–1348. [Google Scholar] [CrossRef]
- Taghavi, S.; van der Lelie, D.; Hoffman, A.; Zhang, Y.-B.; Walla, M.D.; Vangronsveld, J.; Newman, L.; Monchy, S. Genome Sequence of the Plant Growth Promoting Endophytic Bacterium Enterobacter sp. 638. PLoS Genet. 2010, 6, e1000943. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-S.; Ahn, Y.-R.; Song, G.C.; Ghim, S.-Y.; Lee, S.; Lee, G.; Ryu, C.-M. Impact of a Bacterial Volatile 2,3-Butanediol on Bacillus subtilis Rhizosphere Robustness. Front. Microbiol. 2016, 7, 993. [Google Scholar] [CrossRef] [Green Version]
- Ryu, C.-M.; Farag, M.A.; Hu, C.-H.; Reddy, M.S.; Wei, H.-X.; Paré, P.W.; Kloepper, J.W. Bacterial volatiles promote growth in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 4927–4932. [Google Scholar] [CrossRef] [Green Version]
- Spaepen, S.; Vanderleyden, J.; Remans, R. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 2007, 31, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Ismail, M.; Amin, M.; Eid, A.; Hassan, S.; Mahgoub, H.; Lashin, I.; Abdelwahab, A.; Azab, E.; Gobouri, A.; Elkelish, A.; et al. Comparative Study between Exogenously Applied Plant Growth Hormones versus Metabolites of Microbial Endophytes as Plant Growth-Promoting for Phaseolus vulgaris L. Cells 2021, 10, 1059. [Google Scholar] [CrossRef]
- Schmidt, R.; Etalo, D.; De Jager, V.; Gerards, S.; Zweers, H.; de Boer, W.; Garbeva, P. Microbial Small Talk: Volatiles in Fungal–Bacterial Interactions. Front. Microbiol. 2016, 6, 1495. [Google Scholar] [CrossRef] [Green Version]
- Ryjenkov, D.A.; Tarutina, M.; Moskvin, O.; Gomelsky, M. Cyclic Diguanylate Is a Ubiquitous Signaling Molecule in Bacteria: Insights into Biochemistry of the GGDEF Protein Domain. J. Bacteriol. 2005, 187, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Römling, U.; Gomelsky, M.; Galperin, M. C-di-GMP: The dawning of a novel bacterial signalling system. Mol. Microbiol. 2005, 57, 629–639. [Google Scholar] [CrossRef]
- González, G.; Mendoza, F.; Covarrubias, J.; Morán, N.; Acosta, J. Rendimiento y calidad de semilla de frijol en dos épocas de siembra en la región del bajío. Agricult Técn Méx 2008, 34, 421–430. [Google Scholar]
- Castellanos-Ramos, J.; Guzmán-Maldonado, H.; Kelly, J.; Acosta-Gallegos, J. Registration of ‘Flor de Junio Marcela’ common bean. Crop Sci. 2003, 43, 1121–1122. [Google Scholar] [CrossRef]
- De Campos, S.B.; Youn, J.-W.; Farina, R.; Jaenicke, S.; Jünemann, S.; Szczepanowski, R.; Beneduzi, A.; Vargas, L.; Goesmann, A.; Wendisch, V.F.; et al. Changes in Root Bacterial Communities Associated to Two Different Development Stages of Canola (Brassica napus L. var oleifera) Evaluated through Next-Generation Sequencing Technology. Microb. Ecol. 2013, 65, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, W.D. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Drancourt, M.; Bollet, C.; Carlioz, A.; Martelin, R.; Gayral, J.-P.; Raoult, D. 16S Ribosomal DNA Sequence Analysis of a Large Collection of Environmental and Clinical Unidentifiable Bacterial Isolates. J. Clin. Microbiol. 2000, 38, 3623–3630. [Google Scholar] [CrossRef] [Green Version]
- Soergel, D.A.W.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehr, J.P.; McReynolds, L.A. Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl. Environ. Microbiol. 1989, 55, 2522–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarita, S.; Sharma, P.K.; Priefer, U.B.; Prell, J. Direct amplification of rhizobial nodC sequences from soil total DNA and comparison to nodC diversity of root nodule isolates. FEMS Microbiol. Ecol. 2005, 54, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Feng, K.; Wei, Z.; Wang, Z.; Deng, Y. ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies. Int. J. Environ. Res. Public Health 2020, 17, 5958. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Paz, F.; Herrera-Estrella, L.; Heil, M. All Set before Flowering: A 16S Gene Amplicon-Based Analysis of the Root Microbiome Recruited by Common Bean (Phaseolus vulgaris) in Its Centre of Domestication. Plants 2022, 11, 1631. https://doi.org/10.3390/plants11131631
Medina-Paz F, Herrera-Estrella L, Heil M. All Set before Flowering: A 16S Gene Amplicon-Based Analysis of the Root Microbiome Recruited by Common Bean (Phaseolus vulgaris) in Its Centre of Domestication. Plants. 2022; 11(13):1631. https://doi.org/10.3390/plants11131631
Chicago/Turabian StyleMedina-Paz, Francisco, Luis Herrera-Estrella, and Martin Heil. 2022. "All Set before Flowering: A 16S Gene Amplicon-Based Analysis of the Root Microbiome Recruited by Common Bean (Phaseolus vulgaris) in Its Centre of Domestication" Plants 11, no. 13: 1631. https://doi.org/10.3390/plants11131631
APA StyleMedina-Paz, F., Herrera-Estrella, L., & Heil, M. (2022). All Set before Flowering: A 16S Gene Amplicon-Based Analysis of the Root Microbiome Recruited by Common Bean (Phaseolus vulgaris) in Its Centre of Domestication. Plants, 11(13), 1631. https://doi.org/10.3390/plants11131631