
The Pattern Recognition Receptor FLS2 Can Shape the Arabidopsis Rhizosphere Microbiome β-Diversity but Not EFR1 and CERK1

 , and
, and
Abstract
:1. Introduction
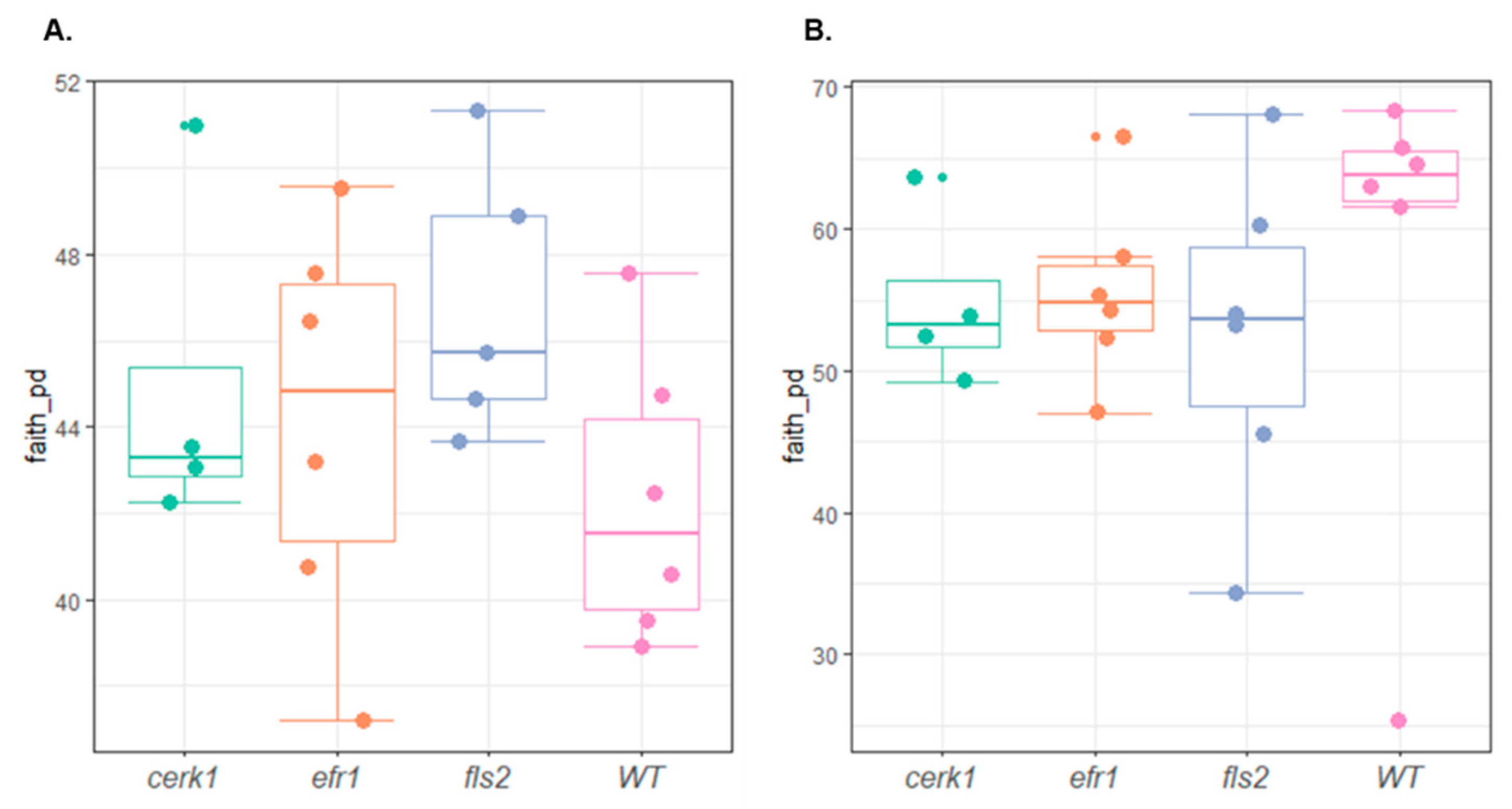
2. Results
2.1. Taxonomic Composition of Bacterial and Fungal Rhizosphere Communities
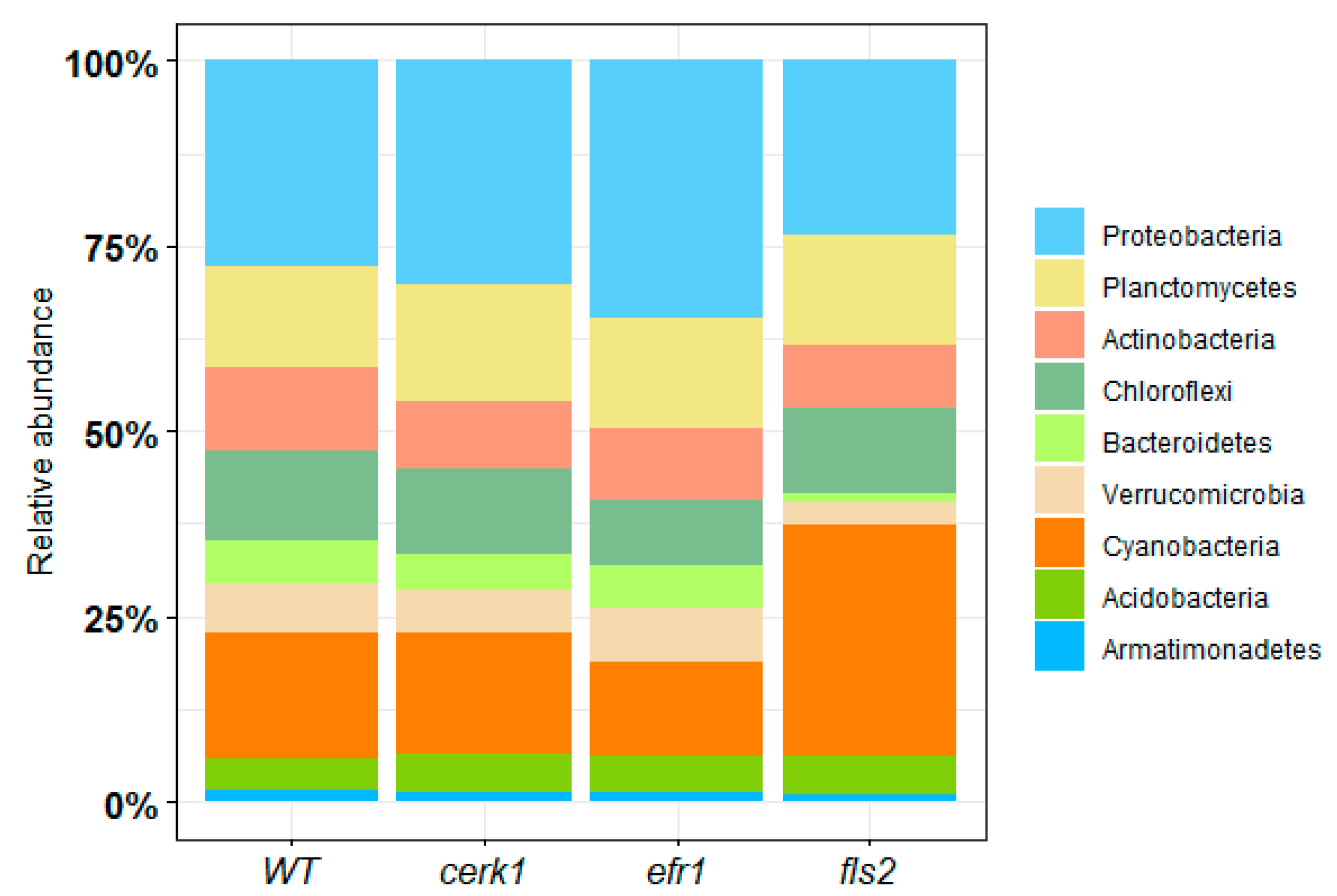
2.1.1. Bacterial Relative Abundance
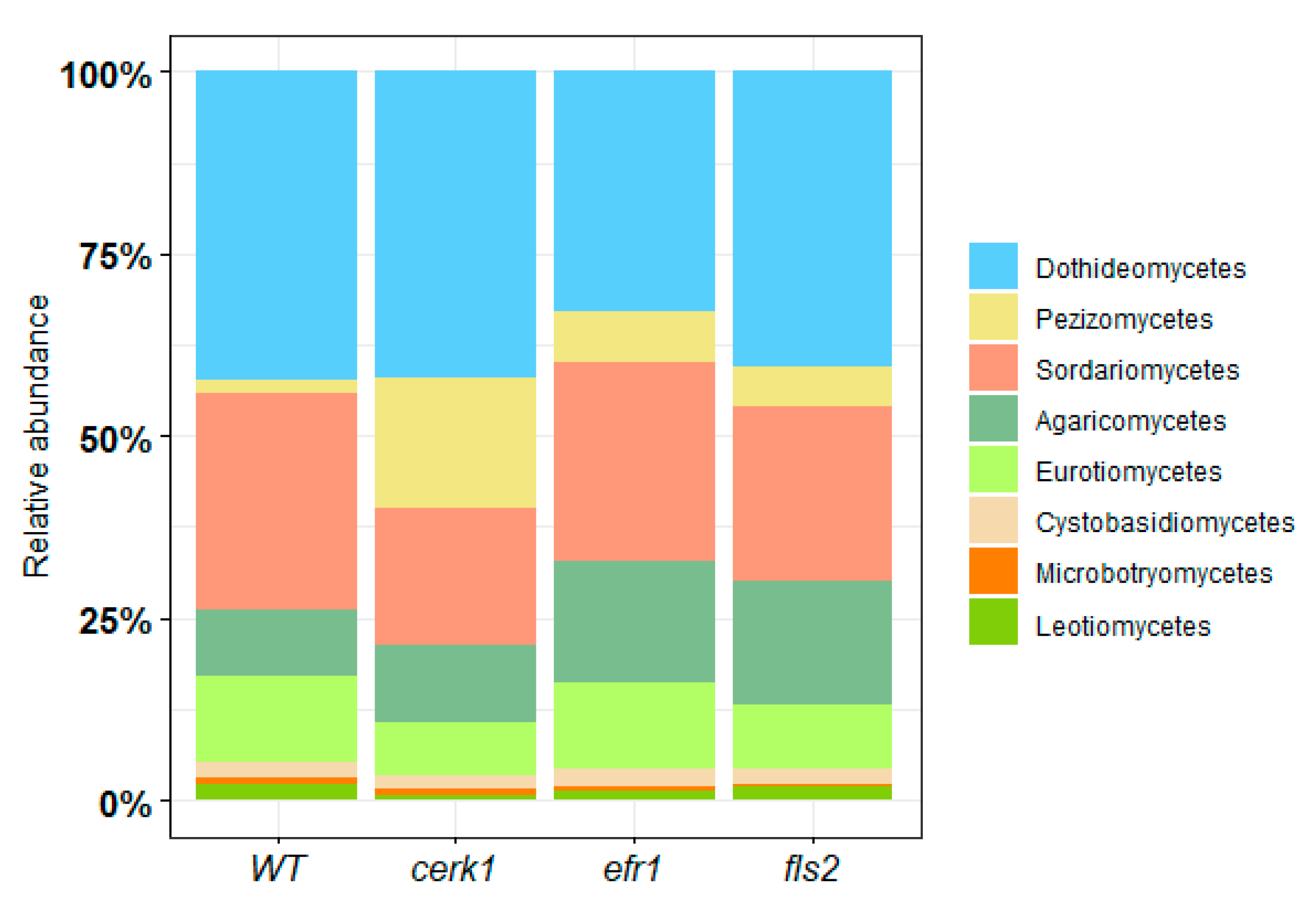
2.1.2. Fungal Relative Abundance
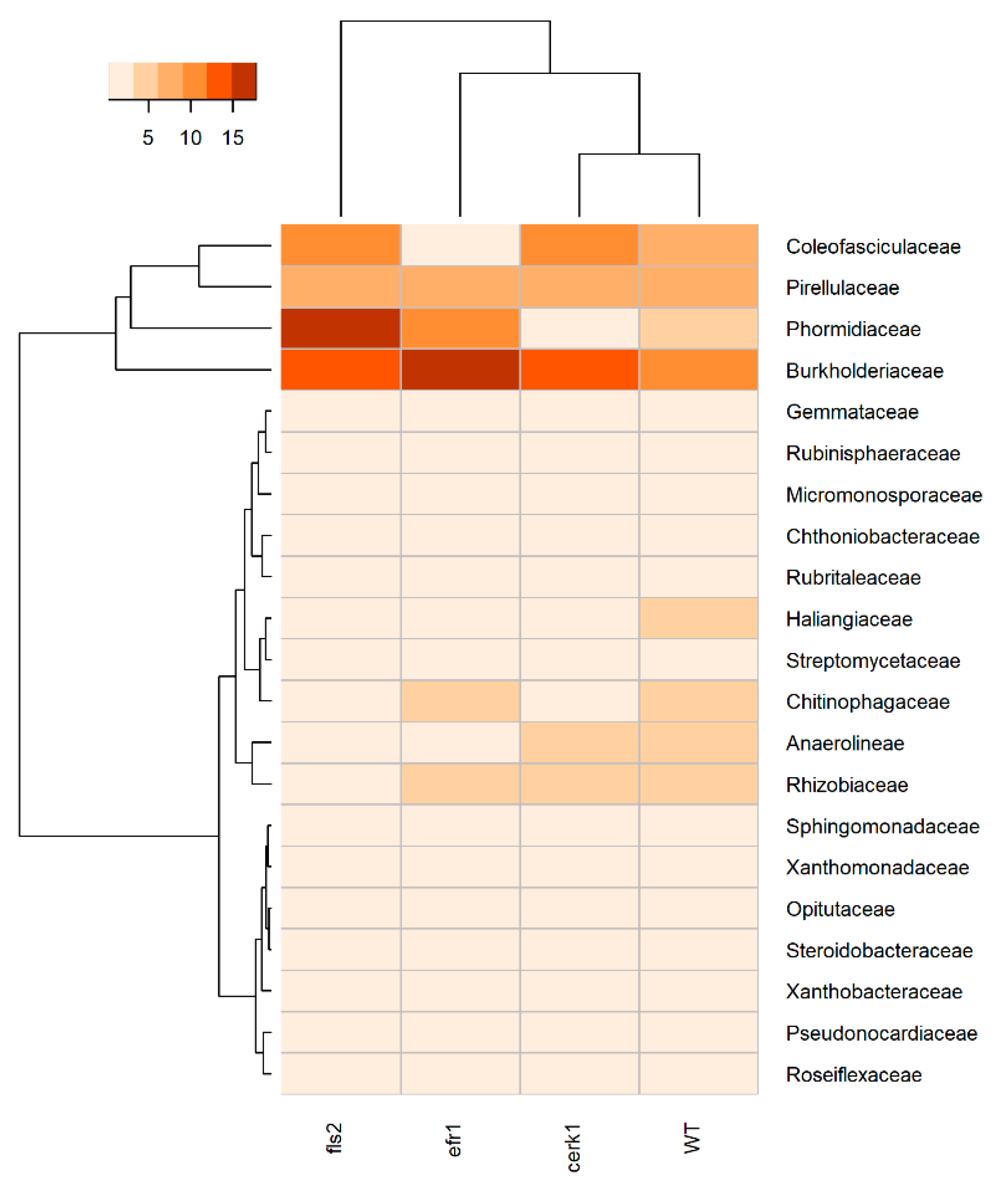
2.2. The Pezizaceae Family Is Highly Enriched in the cerk1 Mutant and All Other Taxonomic Ranks
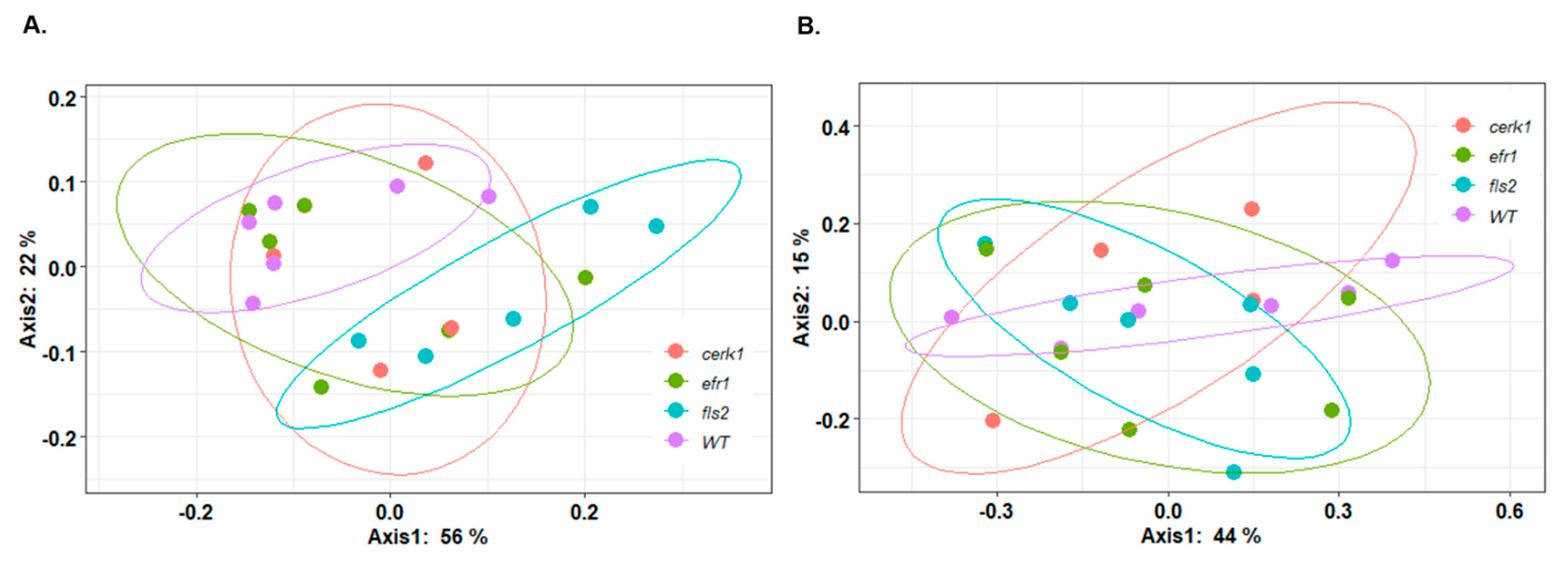
2.3. Compositional Differences in the Microbial Community between Genotypes
3. Discussion
4. Materials and Methods
4.1. Plant and Soil Material Preparation
4.2. Sampling Procedure
4.3. DNA Extraction
4.4. 16S rDNA and ITS Sequencing
4.5. Sequence Processing for 16S and ITS Amplicons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voges MJ EE, E.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, C.H.; Samuel, B.S.; Bush, J.; Ausubel, F.M. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat. Plants 2015, 1, 15051. [Google Scholar] [CrossRef] [Green Version]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; Del Rio, T.G.; Jones, C.D.; Tringe, S.G.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrillo, G.; Teixeira, P.J.; Paredes, S.H. Root microbiota drive direct integration of phosphate stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; Diouf, D.; Kane, A.; Bena, G.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Identification of a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 2020, 96, fiaa067. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 2019, 19, 201. [Google Scholar] [CrossRef]
- Berlanas, C.; Berbegal, M.; Elena, G.; Laidani, M.; Cibriain, J.F.; Sagües, A.; Gramaje, D. The Fungal and Bacterial Rhizosphere Microbiome Associated With Grapevine Rootstock Genotypes in Mature and Young Vineyards. Front. Microbiol. 2019, 10, 1142. [Google Scholar] [CrossRef]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gomez, L.; Bauer, Z.; Boller, T. Both the extracellular leucine-rich repeat domain and the kinase activity of FSL2 are required for flagellin binding and signaling in Arabidopsis. Plant Cell 2001, 13, 1155–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.D.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 2004, 428, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C.; Kunze, G.; Chinchilla, D.; Caniard, A.; Jones, J.D.G.; Boller, T.; Felix, G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 2006, 125, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Miya, A.; Albert, P.; Shinya, T.; Desaki, Y.; Ichimura, K.; Shirasu, K.; Narusaka, Y.; Kawakami, N.; Kaku, H.; Shibuya, N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Natl Acad. Sci. USA 2007, 104, 19613–19618. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, R.H. Evolution and Measurement of Species Diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef] [Green Version]
- Saijo, Y.; Loo, E.P.I.; Yasuda, S. Pattern recognition receptors and signaling in plant-microbe interactions. Plant J. 2018, 93, 592–613. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, A.; Herrera, H.; Charles, T.C.; Arriagada, C. Fungal and Bacterial Microbiome Associated with the Rhizosphere of Native Plants from the Atacama Desert. Microorganisms 2020, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, J.P.; Hoffmann, L.; Cabral, B.C.A.; Dias, V.H.G.; Miranda, M.R.; Azevedo, M.A.C.; Boschiero, C.; Bastos, W.R.; Silva, R. Contrasting the microbiomes from forest rhizosphere and deeper bulk soil from an Amazon rainforest reserve. Gene 2018, 642, 389–397. [Google Scholar] [CrossRef]
- Spatafora, J.W.; Sung, G.H.; Johnson, D.; Hesse, C.; O’Rourke, B.; Serdani, M.; Spotts, R.; Lutzoni, F.; Hofstetter, V.; Miadlikowska, J.; et al. A five-gene phylogenetic analysis of the Pezizomycotina. Mycologia 2006, 98, 1018–1028. [Google Scholar] [CrossRef]
- Uppalapati, S.R.; Young, C.A.; Marek, S.M.; Mysore, K.S. Phymatotrichum (cotton) root rot caused by Phymatotrichopsis omnivora: Retrospects and prospects. Mol. Plant Pathol. 2010, 11, 325–334. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, X.-C.; Neece, D.; Ramonell, K.M.; Clough, S.; Kim, S.-Y.; Stacey, M.G.; Stacey, G. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 2008, 20, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petutschnig, E.K.; Stolze, M.; Lipka, U.; Kopischke, M.; Horlacher, J.; Valerius, O.; Rozhon, W.; Gust, A.A.; Kemmerling, B.; Poppenberger, B.; et al. A novel Arabidopsis CHITIN ELICITOR RECEPTOR KINASE 1 (CERK1) mutant with enhanced pathogen-induced cell death and altered receptor processing. New Phytol. 2014, 204, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Lajunen, H.M.; Erbs, G.; Newman, M.-A.; Kolb, D.; Tsuda, K.; Katagiri, F.; Fliegmann, J.; Bono, J.-J.; Cullimore, J.V.; et al. Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial peptidoglycan sensing and immunity to bacterial infection. Proc. Natl. Acad. Sci. USA 2011, 108, 19824–19829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Ibanez, S.; Hann, D.R.; Ntoukakis, V.; Petutschnig, E.; Lipka, V.; Rathjen, J.P. AvrPtoB targets the LysM receptor kinase CERK1 to promote bacterial virulence on plants. Curr. Biol. CB 2009, 19, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Dastogeer, K.M.G.; Tumpa, F.H.; Sultana, A.; Akter, M.A.; Chakraborty, A. Plant microbiome–an account of the drivers that shape community composition and structure. Curr. Plant Biol. 2020, 23, 100161. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. USA 2018, 115, E1157–E1165. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate Estimation of Fungal Diversity and Abundance through Improved Lineage-Specific Primers Optimized for Illumina Amplicon Sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [Green Version]
- Víquez-R, L.; Fleischer, R.; Wilhelm, K.; Tschapka, M.; Sommer, S. Jumping the green wall: The use of PNA-DNA clamps to enhance microbiome sampling depth in wildlife microbiome research. Ecol. Evol. 2020, 24, 11779–11786. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 27, 1E.5.1–1E.5.20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson AJ, A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Lozupone, C.; Knight, R. Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microbial Community | Genotypes | α-Diversity (Faith) | β-Diversity (Weighted Unifrac) |
|---|---|---|---|
| Bacterial | cerk1 and efr1 | 0.83 | 0.75 |
| cerk1 and fls2 | 0.14 | 0.16 | |
| cerk1 and WT | 0.28 | 0.32 | |
| efr1 and fls2 | 0.36 | 0.07 | |
| efr1 and WT | 0.33 | 0.29 | |
| fls2 and WT | 0.06 | 0.01 | |
| Fungi | cerk1 and efr1 | 0.66 | 0.84 |
| cerk1 and fls2 | 1 | 0.76 | |
| cerk1 and WT | 0.20 | 0.50 | |
| efr1 and fls2 | 0.63 | 0.79 | |
| efr1 and WT | 0.20 | 0.64 | |
| fls2 and WT | 0.20 | 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, J.P.; Lakshmanan, V.; Boschiero, C.; Mysore, K.S. The Pattern Recognition Receptor FLS2 Can Shape the Arabidopsis Rhizosphere Microbiome β-Diversity but Not EFR1 and CERK1. Plants 2022, 11, 1323. https://doi.org/10.3390/plants11101323
Fonseca JP, Lakshmanan V, Boschiero C, Mysore KS. The Pattern Recognition Receptor FLS2 Can Shape the Arabidopsis Rhizosphere Microbiome β-Diversity but Not EFR1 and CERK1. Plants. 2022; 11(10):1323. https://doi.org/10.3390/plants11101323
Chicago/Turabian StyleFonseca, Jose P., Venkatachalam Lakshmanan, Clarissa Boschiero, and Kirankumar S. Mysore. 2022. "The Pattern Recognition Receptor FLS2 Can Shape the Arabidopsis Rhizosphere Microbiome β-Diversity but Not EFR1 and CERK1" Plants 11, no. 10: 1323. https://doi.org/10.3390/plants11101323
APA StyleFonseca, J. P., Lakshmanan, V., Boschiero, C., & Mysore, K. S. (2022). The Pattern Recognition Receptor FLS2 Can Shape the Arabidopsis Rhizosphere Microbiome β-Diversity but Not EFR1 and CERK1. Plants, 11(10), 1323. https://doi.org/10.3390/plants11101323