
Transcription Factor-Based Genetic Engineering in Microalgae



{kind=link}
Abstract
1. Introduction
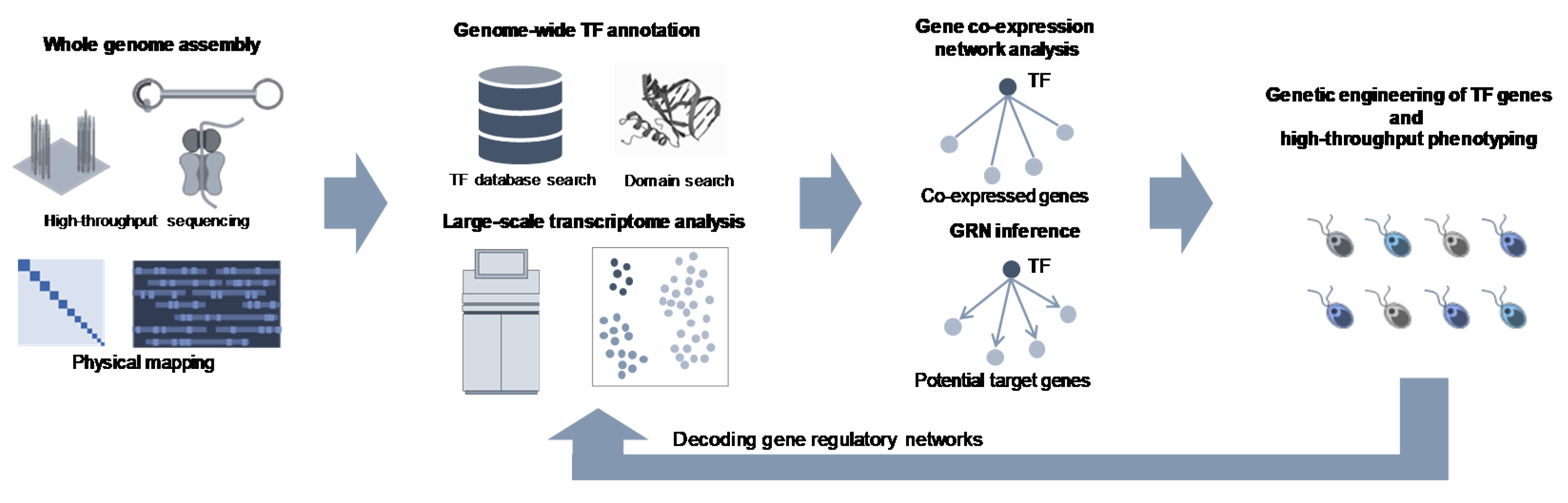
2. Whole Genome Assembly in Microalgae
3. Genome-Wide Identification of TF Repertories in Microalgae
4. Gene Regulatory Networks with TFs
5. TF-based Metabolic Engineering in Microalgae
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Argueso, C.T.; Raines, T.; Kieber, J.J. Cytokinin Signaling and Transcriptional Networks. Curr. Opin. Plant Biol. 2010, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The Transcriptional Regulatory Network in the Drought Response and Its Crosstalk in Abiotic Stress Responses Including Drought, Cold, and Heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Somssich, I.E. Transcriptional Networks in Plant Immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef]
- Ó’Maoiléidigh, D.S.; Graciet, E.; Wellmer, F. Gene Networks Controlling Arabidopsis Thaliana Flower Development. New Phytol. 2014, 201, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, M.; Matus, J.T.; Riechmann, J.L. Genome-Wide Analyses for Dissecting Gene Regulatory Networks in the Shoot Apical Meristem. J. Exp. Bot. 2016, 67, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-Binding Specificities of Plant Transcription Factors and Their Potential to Define Target Genes. Proc. Natl. Acad. Sci. USA 2014, 111, 2367–2372. [Google Scholar] [CrossRef]
- Jing, Y.; Zhang, D.; Wang, X.; Tang, W.; Wang, W.; Huai, J.; Xu, G.; Chen, D.; Li, Y.; Lin, R. Arabidopsis Chromatin Remodeling Factor PICKLE Interacts with Transcription Factor HY5 to Regulate Hypocotyl Cell Elongation. Plant Cell 2013, 25, 242–256. [Google Scholar] [CrossRef]
- Guo, S.; Sun, B.; Looi, L.-S.; Xu, Y.; Gan, E.-S.; Huang, J.; Ito, T. Co-Ordination of Flower Development Through Epigenetic Regulation in Two Model Species: Rice and Arabidopsis. Plant Cell Physiol. 2015, 56, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Romani, F.; Moreno, J.E. Molecular Mechanisms Involved in Functional Macroevolution of Plant Transcription Factors. New Phytol. 2021, 230, 1345–1353. [Google Scholar] [CrossRef]
- Wendrich, J.R.; Yang, B.; Vandamme, N.; Verstaen, K.; Smet, W.; de Velde, C.V.; Minne, M.; Wybouw, B.; Mor, E.; Arents, H.E.; et al. Vascular Transcription Factors Guide Plant Epidermal Responses to Limiting Phosphate Conditions. Science 2020, 370, eaay4970. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Goh, H.; Azodi, C.B.; Krishnamoorthi, S.; Liu, M.-J.; Urano, D. Evolutionarily Conserved Hierarchical Gene Regulatory Networks for Plant Salt Stress Response. Nat. Plants 2021, 7, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yan, W.; Fu, L.-Y.; Kaufmann, K. Architecture of Gene Regulatory Networks Controlling Flower Development in Arabidopsis Thaliana. Nat. Commun. 2018, 9, 4534. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Scossa, F.; Wen, W.; Luo, J.; Yan, J.; Beleggia, R.; Klee, H.J.; Huang, S.; Papa, R.; Fernie, A.R. Domestication of Crop Metabolomes: Desired and Unintended Consequences. Trends Plant Sci. 2021, 26, 650–661. [Google Scholar] [CrossRef]
- Allan, A.C. Domestication: Colour and Flavour Joined by a Shared Transcription Factor. Curr. Biol. 2019, 29, R57–R59. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Yoshida, T.; Sakurai, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. In Silico Analysis of Transcription Factor Repertoire and Prediction of Stress Responsive Transcription Factors in Soybean. DNA Res. 2009, 16, 353–369. [Google Scholar] [CrossRef]
- Mochida, K.; Yoshida, T.; Sakurai, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. In Silico Analysis of Transcription Factor Repertoires and Prediction of Stress-Responsive Transcription Factors from Six Major Gramineae Plants. DNA Res. 2011, 18, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Yoshida, T.; Sakurai, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. TreeTFDB: An Integrative Database of the Transcription Factors from Six Economically Important Tree Crops for Functional Predictions and Comparative and Functional Genomics. DNA Res. 2013, 20, 151–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riaño-Pachón, D.M.; Ruzicic, S.; Dreyer, I.; Mueller-Roeber, B. PlnTFDB: An Integrative Plant Transcription Factor Database. BMC Bioinform. 2007, 8, 42. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a Central Hub for Transcription Factors and Regulatory Interactions in Plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Zemlyanskaya, E.V.; Dolgikh, V.A.; Levitsky, V.G.; Mironova, V. Transcriptional Regulation in Plants: Using Omics Data to Crack the Cis-Regulatory Code. Curr. Opin. Plant Biol. 2021, 63, 102058. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.I.; Kong, S.L.; Akmar Abdullah, S.N.; Munusamy, U. RNA-Seq and ChIP-Seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism. Int. J. Mol. Sci. 2020, 21, 167. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.-Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis Transcription Factors: Genome-Wide Comparative Analysis Among Eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Misumi, O.; Shin-i, T.; Maruyama, S.; Takahara, M.; Miyagishima, S.; Mori, T.; Nishida, K.; Yagisawa, F.; Nishida, K.; et al. Genome Sequence of the Ultrasmall Unicellular Red Alga Cyanidioschyzon Merolae 10D. Nature 2004, 428, 653–657. [Google Scholar] [CrossRef]
- Armbrust, E.V.; Berges, J.A.; Bowler, C.; Green, B.R.; Martinez, D.; Putnam, N.H.; Zhou, S.; Allen, A.E.; Apt, K.E.; Bechner, M.; et al. The Genome of the Diatom Thalassiosira Pseudonana: Ecology, Evolution, and Metabolism. Science 2004, 306, 79–86. [Google Scholar] [CrossRef]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Price, D.C.; Chan, C.X.; Yoon, H.S.; Yang, E.C.; Qiu, H.; Weber, A.P.M.; Schwacke, R.; Gross, J.; Blouin, N.A.; Lane, C.; et al. Cyanophora Paradoxa Genome Elucidates Origin of Photosynthesis in Algae and Plants. Science 2012, 335, 843–847. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Price, D.C.; Chan, C.X.; Qiu, H.; Rose, N.; Ball, S.; Weber, A.P.M.; Cecilia Arias, M.; Henrissat, B.; Coutinho, P.M.; et al. Genome of the Red Alga Porphyridium Purpureum. Nat. Commun. 2013, 4, 1941. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Arriola, M.B.; Velmurugan, N.; Zhang, Y.; Plunkett, M.H.; Hondzo, H.; Barney, B.M. Genome Sequences of Chlorella Sorokiniana UTEX 1602 and Micractinium Conductrix SAG 241.80: Implications to Maltose Excretion by a Green Alga. Plant J. 2018, 93, 566–586. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Building Near-Complete Plant Genomes. Curr. Opin. Plant Biol. 2020, 54, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Acquadro, A.; Portis, E.; Valentino, D.; Barchi, L.; Lanteri, S. “Mind the Gap”: Hi-C Technology Boosts Contiguity of the Globe Artichoke Genome in Low-Recombination Regions. G3 Genes Genomes Genet. 2020, 10, 3557–3564. [Google Scholar] [CrossRef]
- Blaby-Haas, C.E.; Merchant, S.S. Comparative and Functional Algal Genomics. Annu. Rev. Plant Biol. 2019, 70, 605–638. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Hayes, R.D.; Calhoun, S.; Kamel, B.; Wang, A.; Ahrendt, S.; Dusheyko, S.; Nikitin, R.; Mondo, S.J.; Salamov, A.; et al. PhycoCosm, a Comparative Algal Genomics Resource. Nucleic Acids Res. 2021, 49, D1004–D1011. [Google Scholar] [CrossRef]
- Hanschen, E.R.; Starkenburg, S.R. The State of Algal Genome Quality and Diversity. Algal Res. 2020, 50, 101968. [Google Scholar] [CrossRef]
- Roth, M.S.; Cokus, S.J.; Gallaher, S.D.; Walter, A.; Lopez, D.; Erickson, E.; Endelman, B.; Westcott, D.; Larabell, C.A.; Merchant, S.S.; et al. Chromosome-Level Genome Assembly and Transcriptome of the Green Alga Chromochloris Zofingiensis Illuminates Astaxanthin Production. Proc. Natl. Acad. Sci. USA 2017, 114, E4296–E4305. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liang, S.; Zhang, Z.; Liu, H.; Wang, S.; Pan, K.; Xu, J.; Ren, X.; Pei, S.; Yang, G. Genome Assembly of Nannochloropsis Oceanica Provides Evidence of Host Nucleus Overthrow by the Symbiont Nucleus during Speciation. Commun. Biol. 2019, 2, 249. [Google Scholar] [CrossRef] [PubMed]
- Kouzai, Y.; Shimizu, M.; Inoue, K.; Uehara-Yamaguchi, Y.; Takahagi, K.; Nakayama, R.; Matsuura, T.; Mori, I.C.; Hirayama, T.; Abdelsalam, S.S.H.; et al. BdWRKY38 Is Required for the Incompatible Interaction of Brachypodium Distachyon with the Necrotrophic Fungus Rhizoctonia Solani. Plant J. 2020, 104, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, X.; Liu, Q.; Ahammed, G.J.; Lin, R.; Wang, L.; Shao, S.; Yu, J.; Zhou, Y. The HY5 and MYB15 Transcription Factors Positively Regulate Cold Tolerance in Tomato via the CBF Pathway. Plant Cell Environ. 2020, 43, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Watt, C.; Zhou, G.; Li, C. Harnessing Transcription Factors as Potential Tools to Enhance Grain Size Under Stressful Abiotic Conditions in Cereal Crops. Front. Plant Sci. 2020, 11, 1273. [Google Scholar] [CrossRef]
- Coego, A.; Brizuela, E.; Castillejo, P.; Ruíz, S.; Koncz, C.; del Pozo, J.C.; Piñeiro, M.; Jarillo, J.A.; Paz-Ares, J.; León, J.; et al. The TRANSPLANTA Collection of Arabidopsis Lines: A Resource for Functional Analysis of Transcription Factors Based on Their Conditional Overexpression. Plant J. 2014, 77, 944–953. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Z.; Zhang, Q.; Liu, Y.; Zhu, B.; Cao, J.; Li, Z.; Han, L.; Jia, J.; Zhao, G.; et al. Generation of Wheat Transcription Factor FOX Rice Lines and Systematic Screening for Salt and Osmotic Stress Tolerance. PLoS ONE 2015, 10, e0132314. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mizukado, S.; Fujita, Y.; Ichikawa, T.; Nakazawa, M.; Seki, M.; Matsui, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Identification of Stress-Tolerance-Related Transcription-Factor Genes via Mini-Scale Full-Length CDNA Over-EXpressor (FOX) Gene Hunting System. Biochem. Biophys. Res. Commun. 2007, 364, 250–257. [Google Scholar] [CrossRef]
- Mitsuda, N.; Ikeda, M.; Takada, S.; Takiguchi, Y.; Kondou, Y.; Yoshizumi, T.; Fujita, M.; Shinozaki, K.; Matsui, M.; Ohme-Takagi, M. Efficient Yeast One-/Two-Hybrid Screening Using a Library Composed Only of Transcription Factors in Arabidopsis Thaliana. Plant Cell Physiol. 2010, 51, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Yin, K.-Q.; Liu, S.-N.; Yang, Y.; Gu, T.; Wing Hui, J.M.; Zhang, L.; Miao, J.; Kondou, Y.; Matsui, M.; et al. A High-Throughput Screening System for Arabidopsis Transcription Factors and Its Application to Med25-Dependent Transcriptional Regulation. Mol. Plant 2011, 4, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Riaño-Pachón, D.M.; Corrêa, L.G.G.; Trejos-Espinosa, R.; Mueller-Roeber, B. Green Transcription Factors: A Chlamydomonas Overview. Genetics 2008, 179, 31–39. [Google Scholar] [CrossRef]
- Thiriet-Rupert, S.; Carrier, G.; Chénais, B.; Trottier, C.; Bougaran, G.; Cadoret, J.-P.; Schoefs, B.; Saint-Jean, B. Transcription Factors in Microalgae: Genome-Wide Prediction and Comparative Analysis. BMC Genomics 2016, 17, 282. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Dixon, R.A. Co-Expression Networks for Plant Biology: Why and How. Acta Biochim. Biophys. Sin. 2019, 51, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.Y.; Sugiyama, K.; Sawada, Y.; Tohge, T.; Obayashi, T.; Suzuki, A.; Araki, R.; Sakurai, N.; Suzuki, H.; Aoki, K.; et al. Omics-Based Identification of Arabidopsis Myb Transcription Factors Regulating Aliphatic Glucosinolate Biosynthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6478–6483. [Google Scholar] [CrossRef]
- Righetti, K.; Vu, J.L.; Pelletier, S.; Vu, B.L.; Glaab, E.; Lalanne, D.; Pasha, A.; Patel, R.V.; Provart, N.J.; Verdier, J.; et al. Inference of Longevity-Related Genes from a Robust Coexpression Network of Seed Maturation Identifies Regulators Linking Seed Storability to Biotic Defense-Related Pathways. Plant Cell 2015, 27, 2692–2708. [Google Scholar] [CrossRef]
- Obayashi, T.; Aoki, Y.; Tadaka, S.; Kagaya, Y.; Kinoshita, K. ATTED-II in 2018: A Plant Coexpression Database Based on Investigation of the Statistical Property of the Mutual Rank Index. Plant Cell Physiol. 2018, 59, e3. [Google Scholar] [CrossRef]
- Aoki, Y.; Okamura, Y.; Ohta, H.; Kinoshita, K.; Obayashi, T. ALCOdb: Gene Coexpression Database for Microalgae. Plant Cell Physiol. 2016, 57, e3. [Google Scholar] [CrossRef]
- Hansen, B.O.; Meyer, E.H.; Ferrari, C.; Vaid, N.; Movahedi, S.; Vandepoele, K.; Nikoloski, Z.; Mutwil, M. Ensemble Gene Function Prediction Database Reveals Genes Important for Complex I Formation in Arabidopsis Thaliana. New Phytol. 2018, 217, 1521–1534. [Google Scholar] [CrossRef]
- Romero-Campero, F.J.; Perez-Hurtado, I.; Lucas-Reina, E.; Romero, J.M.; Valverde, F. ChlamyNET: A Chlamydomonas Gene Co-Expression Network Reveals Global Properties of the Transcriptome and the Early Setup of Key Co-Expression Patterns in the Green Lineage. BMC Genomics 2016, 17, 227. [Google Scholar] [CrossRef] [PubMed]
- Salomé, P.A.; Merchant, S.S. Co-Expression Networks in Chlamydomonas Reveal Significant Rhythmicity in Batch Cultures and Empower Gene Function Discovery. Plant Cell 2021, 33, 1058–1082. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Salomé, P.A.; Merchant, S.S.; Pellegrini, M. Single-Cell RNA Sequencing of Batch Chlamydomonas Cultures Reveals Heterogeneity in Their Diurnal Cycle Phase. Plant Cell 2021, 33, 1042–1057. [Google Scholar] [CrossRef]
- Mochida, K.; Koda, S.; Inoue, K.; Nishii, R. Statistical and Machine Learning Approaches to Predict Gene Regulatory Networks From Transcriptome Datasets. Front. Plant Sci. 2018, 9, 1770. [Google Scholar] [CrossRef]
- Zhang, R.; Patena, W.; Armbruster, U.; Gang, S.S.; Blum, S.R.; Jonikas, M.C. High-Throughput Genotyping of Green Algal Mutants Reveals Random Distribution of Mutagenic Insertion Sites and Endonucleolytic Cleavage of Transforming DNA. Plant Cell 2014, 26, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Patena, W.; Fauser, F.; Jinkerson, R.E.; Saroussi, S.; Meyer, M.T.; Ivanova, N.; Robertson, J.M.; Yue, R.; Zhang, R.; et al. A Genome-Wide Algal Mutant Library and Functional Screen Identifies Genes Required for Eukaryotic Photosynthesis. Nat. Genet. 2019, 51, 627–635. [Google Scholar] [CrossRef]
- Gargouri, M.; Park, J.-J.; Holguin, F.O.; Kim, M.-J.; Wang, H.; Deshpande, R.R.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. Identification of Regulatory Network Hubs That Control Lipid Metabolism in Chlamydomonas Reinhardtii. J. Exp. Bot. 2015, 66, 4551–4566. [Google Scholar] [CrossRef]
- Valledor, L.; Furuhashi, T.; Recuenco-Muñoz, L.; Wienkoop, S.; Weckwerth, W. System-Level Network Analysis of Nitrogen Starvation and Recovery in Chlamydomonas Reinhardtii Reveals Potential New Targets for Increased Lipid Accumulation. Biotechnol. Biofuels 2014, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Xie, X.; Wu, M.; Lin, Z.; Yin, J.; Lou, S.; Huang, Y.; Hu, Z. Understanding the Functions of Endogenous DOF Transcript Factor in Chlamydomonas Reinhardtii. Biotechnol. Biofuels 2019, 12, 67. [Google Scholar] [CrossRef]
- Kim, H.U. Lipid Metabolism in Plants. Plants 2020, 9, 871. [Google Scholar] [CrossRef] [PubMed]
- Bajhaiya, A.K.; Dean, A.P.; Zeef, L.A.H.; Webster, R.E.; Pittman, J.K. PSR1 Is a Global Transcriptional Regulator of Phosphorus Deficiency Responses and Carbon Storage Metabolism in Chlamydomonas Reinhardtii. Plant Physiol. 2016, 170, 1216–1234. [Google Scholar] [CrossRef]
- Hidayati, N.A.; Yamada-Oshima, Y.; Iwai, M.; Yamano, T.; Kajikawa, M.; Sakurai, N.; Suda, K.; Sesoko, K.; Hori, K.; Obayashi, T.; et al. Lipid Remodeling Regulator 1 (LRL1) Is Differently Involved in the Phosphorus-Depletion Response from PSR1 in Chlamydomonas Reinhardtii. Plant J. 2019, 100, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Shin, S.; Choi, B.Y.; Kim, H.; Jang, S.; Kajikawa, M.; Yamano, T.; Kong, F.; Légeret, B.; Fukuzawa, H.; et al. The BZIP1 Transcription Factor Regulates Lipid Remodeling and Contributes to ER Stress Management in Chlamydomonas Reinhardtii. Plant Cell 2019, 31, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Ajjawi, I.; Verruto, J.; Aqui, M.; Soriaga, L.B.; Coppersmith, J.; Kwok, K.; Peach, L.; Orchard, E.; Kalb, R.; Xu, W.; et al. Lipid Production in Nannochloropsis Gaditana Is Doubled by Decreasing Expression of a Single Transcriptional Regulator. Nat. Biotechnol. 2017, 35, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, S.; Hirose, Y.; Kanesaki, Y.; Higuchi, S.; Fujiwara, T.; Onuma, R.; Era, A.; Ohbayashi, R.; Uzuka, A.; Nozaki, H.; et al. Acidophilic Green Algal Genome Provides Insights into Adaptation to an Acidic Environment. Proc. Natl. Acad. Sci. USA 2017, 114, E8304–E8313. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, C.; Zhang, K.; He, Y.; Zhao, X.; Yang, L.; Zheng, Z.; Ma, X.; Wang, X.; Wang, W.; et al. Adaptation to Extreme Antarctic Environments Revealed by the Genome of a Sea Ice Green Alga. Curr. Biol. 2020, 30, 3330–3341.e7. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, L.; Salek, M.M.; Trampe, E.L.; Fernandez, V.I.; Lee, K.S.; Kühl, M.; Stocker, R. PhenoChip: A Single-Cell Phenomic Platform for High-Throughput Photophysiological Analyses of Microalgae. Sci. Adv. 2020, 6, eabb2754. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mochdia, K.; Tamaki, S. Transcription Factor-Based Genetic Engineering in Microalgae. Plants 2021, 10, 1602. https://doi.org/10.3390/plants10081602
Mochdia K, Tamaki S. Transcription Factor-Based Genetic Engineering in Microalgae. Plants. 2021; 10(8):1602. https://doi.org/10.3390/plants10081602
Chicago/Turabian StyleMochdia, Keiichi, and Shun Tamaki. 2021. "Transcription Factor-Based Genetic Engineering in Microalgae" Plants 10, no. 8: 1602. https://doi.org/10.3390/plants10081602
APA StyleMochdia, K., & Tamaki, S. (2021). Transcription Factor-Based Genetic Engineering in Microalgae. Plants, 10(8), 1602. https://doi.org/10.3390/plants10081602