
Transcriptome and Metabolome Integrated Analysis of Two Ecotypes of Tetrastigma hemsleyanum Reveals Candidate Genes Involved in Chlorogenic Acid Accumulation


Abstract
1. Introduction
2. Results
2.1. Transcriptome Analysis in RG and SG13 Leaves
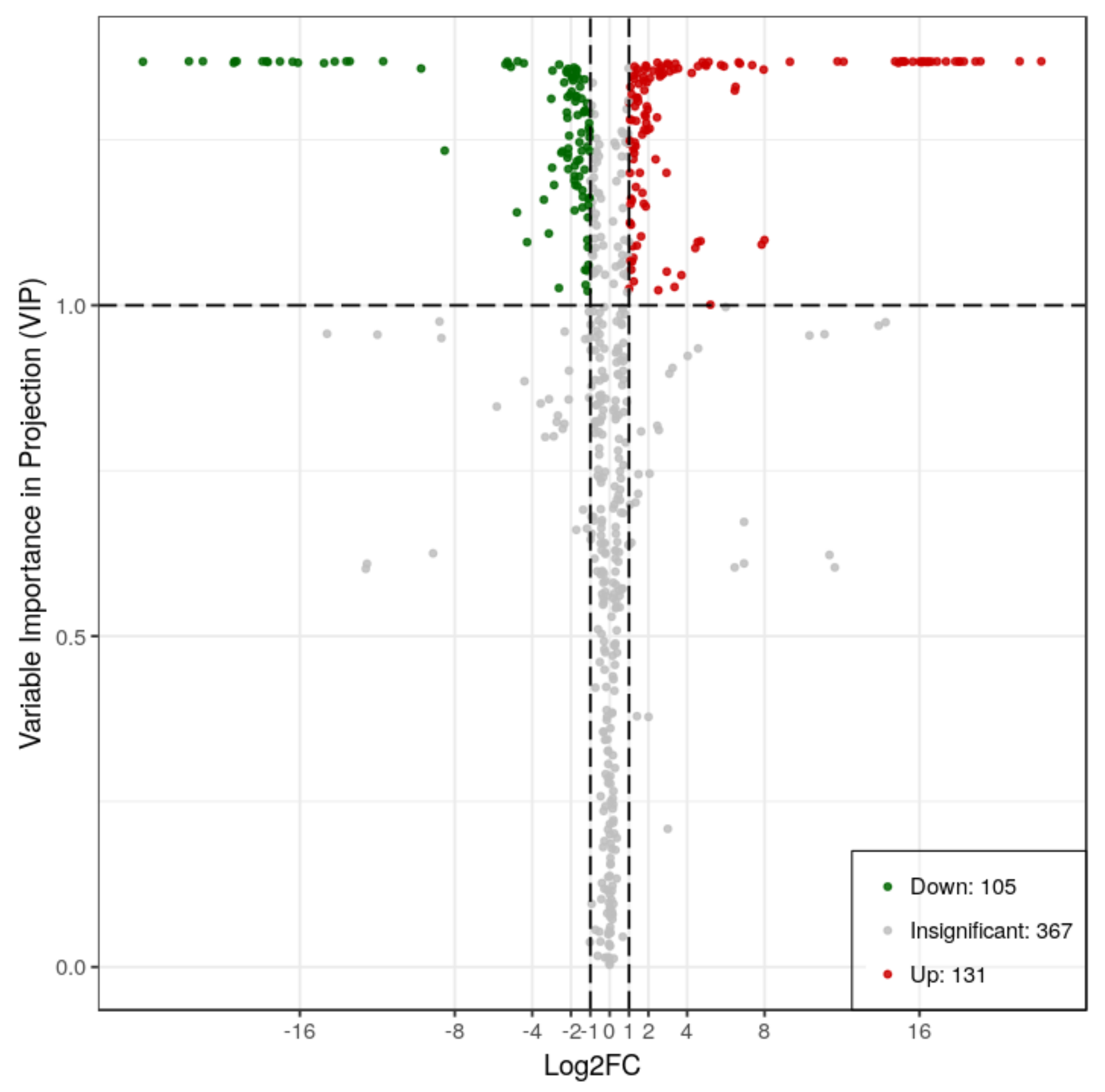
2.2. Metabolic Differences in RG and SG13 Leaves
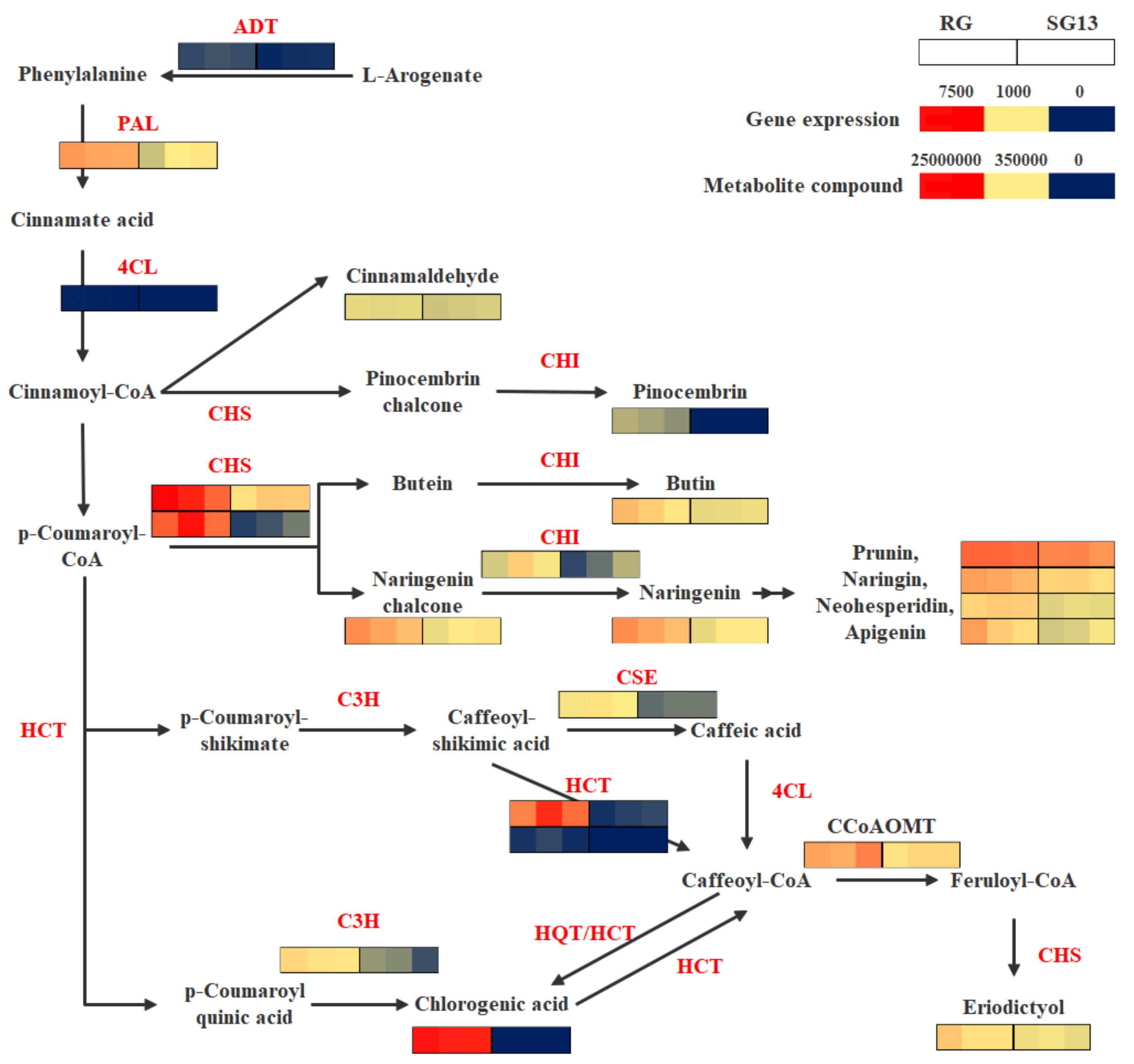
2.3. Integrated Analysis of Metabolites and Transcripts in the Phenylpropanoid and Flavonoid Biosynthesis Pathway
2.4. Validation of Expression of Genes Involved in Chlorogenic Acid Accumulation
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA Isolation and Transcriptome Sequencing
4.3. Metabolic Profiling
4.4. Verification of Candidate Genes by Quantitative Real-Time PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.H.; Jiang, W.M.; Comes, H.P.; Hu, F.S.; Qiu, Y.X.; Fu, C.X. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): Insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. N. Phytol. 2015, 206, 852–867. [Google Scholar] [CrossRef]
- Li, C. Flora of China; Science Press: Beijing, China, 1998; Volume 48, p. 122. [Google Scholar]
- Ji, T.; Ji, W.; Wang, J.; Chen, H.; Peng, X.; Cheng, K.; Qiu, D.; Yang, W. A comprehensive review on traditional uses, chemical compositions, pharmacology properties and toxicology of Tetrastigma hemsleyanum. J. Ethnopharmacol. 2021, 264, 113247. [Google Scholar] [CrossRef]
- Ru, Y.; Chen, X.; Wang, J.; Guo, L.; Lin, Z.; Peng, X.; Qiu, B. Polysaccharides from Tetrastigma hemsleyanum Diels et Gilg: Extraction optimization, structural characterizations, antioxidant and antihyperlipidemic activities in hyperlipidemic mice. Int. J. Biol. Macromol. 2019, 125, 1033–1041. [Google Scholar] [CrossRef]
- Hossain, M.A.; Shah, M.D.; Gnanaraj, C.; Iqbal, M. In vitro total phenolics, flavonoids contents and antioxidant activity of essential oil, various organic extracts from the leaves of tropical medicinal plant Tetrastigma from Sabah. Asian Pac. J. Trop. Med. 2011, 4, 717–721. [Google Scholar] [CrossRef]
- Liu, D.; Yang, J. Studies on the chemical constituents of Tetrastigma hemsleyanum Diels et Gilg Native to China. China J. Chin. Mater. Med. 1999, 24, 611–638. [Google Scholar]
- Zhou, J.; Hao, W.; Lin, X.; Fan, D.; Feng, Z. Antitumor activity of total flavonoids from Tetrastigma hemsleyanum Diels et Gilg is associated with the inhibition of regulatory T cells in mice. Oncol. Targets Ther. 2014, 7, 947–956. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falcone-Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Crozier, T.W.M.; Stalmach, A.; Lean, M.E.J.; Crozier, A. Espresso coffees, caffeine and chlorogenic acid intake: Potential health implications. Food Funct. 2012, 3, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Z.; Li, Y.; Liu, Y.; Cai, W.; Li, C.; Lu, J.; Qiao, Y. A strategy for comprehensive identification of sequential constituents using ultra-high-performance liquid chromatography coupled with linear ion trap–Orbitrap mass spectrometer, application study on chlorogenic acids in Flos Lonicerae japonicae. Talanta 2016, 147, 16–27. [Google Scholar] [CrossRef]
- Clifford, M.N.; Jaganath, I.B.; Ludwig, I.A.; Crozier, A. Chlorogenic acids and the acyl-quinic acids: Discovery, biosynthesis, bioavailability and bioactivity. Nat. Prod. Rep. 2017, 34, 1391–1421. [Google Scholar] [CrossRef]
- Peng, X.; Wu, H.; Chen, H.; Zhang, Y.; Qiu, D.; Zhang, Z. Transcriptome profiling reveals candidate flavonol-related genes of Tetrastigma hemsleyanum under cold stress. BMC Genomics 2019, 20, 687. [Google Scholar] [CrossRef]
- Yan, J.; Qian, L.; Zhu, W.; Qiu, J.; Lu, Q.; Wang, X.; Wu, Q.; Ruan, S.; Huang, Y. Integrated analysis of the transcriptome and metabolome of purple and green leaves of Tetrastigma hemsleyanum reveals gene expression patterns involved in anthocyanin biosynthesis. PLoS ONE 2020, 15, e0230154. [Google Scholar] [CrossRef]
- Peng, X.; Ji, Q.; Fan, S.; Zhang, Y.; Zhang, J. Genetic diversity in populations of the endangered medicinal plant Tetrastigma hemsleyanum revealed by ISSR and SRAP markers: Implications for conservation. Genet. Resour. Crop Evol. 2015, 62, 1069–1078. [Google Scholar] [CrossRef]
- Lin, W.; Chai, Q.; Wang, W.; Li, Y.; Qiu, B.; Peng, X.; Lai, T.; Huang, J.; Lin, Z. A novel method for geographical origin identification of Tetrastigma hemsleyanum (Sanyeqing) by near-infrared spectroscopy. Anal. Methods 2018, 10, 2980–2988. [Google Scholar] [CrossRef]
- Tohge, T.; Nishiyama, Y.; Hirai, M.Y.; Yano, M.; Nakajima, J.; Awazuhara, M.; Inoue, E.; Takahashi, H.; Goodenowe, D.B.; Kitayama, M.; et al. Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 2005, 42, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Pollet, B.; Thévenin, J.; Sibout, R.; Denoue, D.; Barrière, Y.; Lapierre, C.; Jouanin, L. Both caffeoyl Coenzyme A 3-O-methyltransferase 1 and caffeic acid O-methyltransferase 1 are involved in redundant functions for lignin, flavonoids and sinapoyl malate biosynthesis in Arabidopsis. Planta 2007, 226, 1117–1129. [Google Scholar] [CrossRef]
- Yin, Y.; Hou, J.; Tian, S.; Yang, L.; Zhang, Z.; Li, W.; Liu, Y. Overexpressing chalcone synthase (CHS) gene enhanced flavonoids accumulation in Glycyrrhiza uralensis hairy roots. Bot. Lett. 2020, 167, 219–231. [Google Scholar] [CrossRef]
- Muir, S.R.; Collins, G.J.; Robinson, S.; Hughes, S.; Bovy, A.; Ric De Vos, C.H.; van Tunen, A.J.; Verhoeyen, M.E. Overexpression of petunia chalcone isomerase in tomato results in fruit containing increased levels of flavonols. Nat. Biotechnol. 2001, 19, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Veghar, H.; Muhammad, A.; Asghar, A.K.; Ghulam, J.K.; Muhammad, S.; Fawwad, A.; Daryoush, B.; Xia, F.; Faezeh, M.; et al. Chlorogenic acid (CGA): A pharmacological review and call for further research. Biomed. Pharmacother. 2018, 97, 67–74. [Google Scholar] [CrossRef]
- Mhlongo, M.I.; Steenkamp, P.A.; Piater, L.A.; Madala, N.E.; Dubery, I.A. Profiling of altered metabolomic states in Nicotiana tabacum cells induced by priming agents. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, E.; Weinert, C.H.; Egert, B.; Trierweiler, B.; Schmidt-Heydt, M.; Horneburg, B.; Graeff-Hönninger, S.; Kulling, S.E.; Geisen, R. Chlorogenic acid, a metabolite identified by untargeted metabolome analysis in resistant tomatoes, inhibits the colonization by Alternaria alternata by inhibiting alternariol biosynthesis. Eur. J. Plant Pathol. 2014, 139, 735–747. [Google Scholar] [CrossRef]
- Felton, G.W.; Workman, J.; Duffey, S.S. Avoidance of antinutritive plant defense: Role of midgut pH in Colorado potato beetle. J. Chem. Ecol. 1992, 18, 571–583. [Google Scholar] [CrossRef]
- Martinez, V.; Mestre, T.C.; Rubio, F.; Girones-Vilaplana, A.; Moreno, D.A.; Mittler, R.; Rivero, R.M. Accumulation of flavonols over hydroxycinnamic acids favors oxidative damage protection under abiotic stress. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Kumar, R.; Vashisth, D.; Misra, A.; Akhtar, M.Q.; Jalil, S.U.; Shanker, K.; Gupta, M.M.; Rout, P.K.; Gupta, A.K.; Shasany, A.K. RNAi down-regulation of cinnamate-4-hydroxylase increases artemisinin biosynthesis in Artemisia annua. Sci. Rep. 2016, 6, 26458. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Stout, J.; Weng, J.; Humphreys, J.; Ruegger, M.O.; Chapple, C. Mutations in the cinnamate 4-hydroxylase gene impact metabolism, growth and development in Arabidopsis. Plant J. 2009, 60, 771–782. [Google Scholar] [CrossRef]
- Chen, W.; Wang, W.; Peng, M.; Gong, L.; Gao, Y.; Wan, J.; Wang, S.; Shi, L.; Zhou, B.; Li, Z.; et al. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 2016, 7, 12767. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Class | RG | SG13 | VIP | Log2 (Fold_Change) |
|---|---|---|---|---|---|
| Quinic acid O-glucuronic acid | Quinate and its derivatives | 31,266,667 | 9 | 1.3688 | −21.73 |
| Chlorogenic acid (3-O-Caffeoylquinic acid) | Quinate and its derivatives | 18,966,667 | 9 | 1.3688 | −21.01 |
| Isovitexin 7-O-glucoside (Saponarin) | Others | 6,086,667 | 9 | 1.3688 | −19.37 |
| C-hexosyl-apigenin O-pentoside | Flavone C-glycosides | 5,670,000 | 9 | 1.3688 | −19.26 |
| C-hexosyl-apigenin O-hexosyl-O-hexoside | Flavone C-glycosides | 2,243,333 | 9 | 1.3687 | −17.93 |
| Kaempferol 3-O-rhamnoside (Kaempferin) | Flavonol | 1,956,667 | 9 | 1.3687 | −17.73 |
| p-Coumaroyl quinic acid O-glucuronic acid | Quinate and its derivatives | 9 | 300,000 | 1.3687 | 15.02 |
| Chrysin 5-O-glucoside (Toringin) | Flavone | 98,967 | 9 | 1.3688 | −13.42 |
| Tricin 4′-O-(syringyl alcohol) ether 5-O-hexoside | Flavonolignan | 29,900 | 9 | 1.3687 | −11.70 |
| Acetyl tryptophan | Amino acid derivatives | 16,166,667 | 601,000 | 1.3688 | −4.75 |
| Accession Number | Function Prediction | KEGG ID | RG Count | SG13 Count | Fold Change (log2FC) | |
|---|---|---|---|---|---|---|
| Transcripts up-regulated in RG | c96568.graph_c0 | ADT | K05359 | 144 | 41 | 1.58 |
| c103381.graph_c0 | PAL | K10775 | 2866 | 805 | 1.63 | |
| c29476.graph_c0 | 4CL | K01904 | 12 | 0 | 3.08 | |
| c94054.graph_c0 | C3H | K09754 | 1166 | 350 | 1.46 | |
| c100085.graph_c0 | CSE | K18368 | 864 | 334 | 1.14 | |
| c100482.graph_c0 | HCT | K13065 | 4779 | 118 | 5.04 | |
| c78409.graph_c0 | HCT | K13065 | 95 | 0 | 6.16 | |
| c102511.graph_c0 | CCoAOMT | K00588 | 3104 | 1280 | 1.06 | |
| c100230.graph_c0 | CHS | K00660 | 6157 | 1532 | 1.80 | |
| c90950.graph_c0 | CHS | K00660 | 5452 | 225 | 4.32 | |
| c102872.graph_c0 | CHI | K01859 | 995 | 341 | 1.31 | |
| Index | Metabolite name | KEGG ID | RG Content | SG13 Content | Fold Change (Log2FC) | |
| Metabolites up-regulated in RG | pme0398 | Chlorogenic acid | C00852 | 18,966,667 | 9 | 21.00704 |
| pme0424 | trans-cinnamaldehyde | C00903 | 94,433 | 45,900 | 1.040802 | |
| pme2979 | Pinocembrin | C09827 | 7690 | 9 | 9.738843 | |
| pme3473 | Butin | C09614 | 516,000 | 114,000 | 2.178337 | |
| pme2960 | Naringenin chalcone | C06561 | 1,218,667 | 225,667 | 2.433038 | |
| pme0377 | Naringenin | C00509 | 1,182,333 | 209,133 | 2.499142 | |
| pme0002 | Neohesperidin | C09806 | 487,000 | 95,667 | 2.347834 | |
| pme0330 | Naringin | C09789 | 1,046,667 | 410,333 | 1.350934 | |
| pme0371 | Prunin | C09099 | 3,910,000 | 1,950,000 | 1.003694 | |
| pme0379 | Apigenin | C01477 | 750,000 | 102,367 | 2.873145 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, S.; Cui, H.; Zhang, L.; Yan, J.; Qian, L.; Ruan, S. Transcriptome and Metabolome Integrated Analysis of Two Ecotypes of Tetrastigma hemsleyanum Reveals Candidate Genes Involved in Chlorogenic Acid Accumulation. Plants 2021, 10, 1288. https://doi.org/10.3390/plants10071288
Yin S, Cui H, Zhang L, Yan J, Qian L, Ruan S. Transcriptome and Metabolome Integrated Analysis of Two Ecotypes of Tetrastigma hemsleyanum Reveals Candidate Genes Involved in Chlorogenic Acid Accumulation. Plants. 2021; 10(7):1288. https://doi.org/10.3390/plants10071288
Chicago/Turabian StyleYin, Shuya, Hairui Cui, Le Zhang, Jianli Yan, Lihua Qian, and Songlin Ruan. 2021. "Transcriptome and Metabolome Integrated Analysis of Two Ecotypes of Tetrastigma hemsleyanum Reveals Candidate Genes Involved in Chlorogenic Acid Accumulation" Plants 10, no. 7: 1288. https://doi.org/10.3390/plants10071288
APA StyleYin, S., Cui, H., Zhang, L., Yan, J., Qian, L., & Ruan, S. (2021). Transcriptome and Metabolome Integrated Analysis of Two Ecotypes of Tetrastigma hemsleyanum Reveals Candidate Genes Involved in Chlorogenic Acid Accumulation. Plants, 10(7), 1288. https://doi.org/10.3390/plants10071288