
A More Accessible, Time-Saving, and Efficient Method for In Vitro Plant Regeneration from Potato Protoplasts


 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
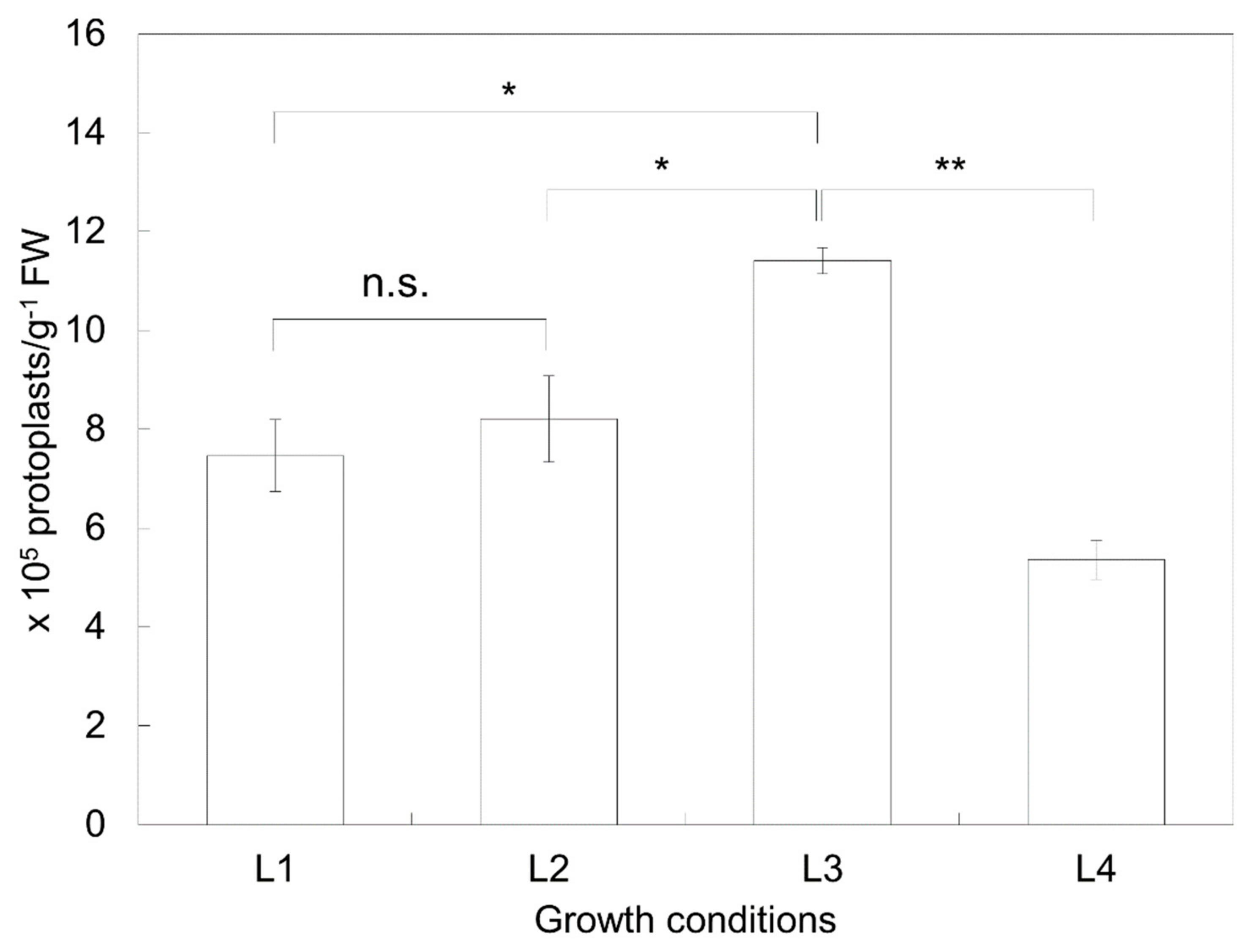
2.1. Isolation Efficiency of Protoplasts Depends on the Preparation Conditions of the Plant Material
2.2. The Isolation Efficiency of Protoplasts Depends on the Extraction Conditions
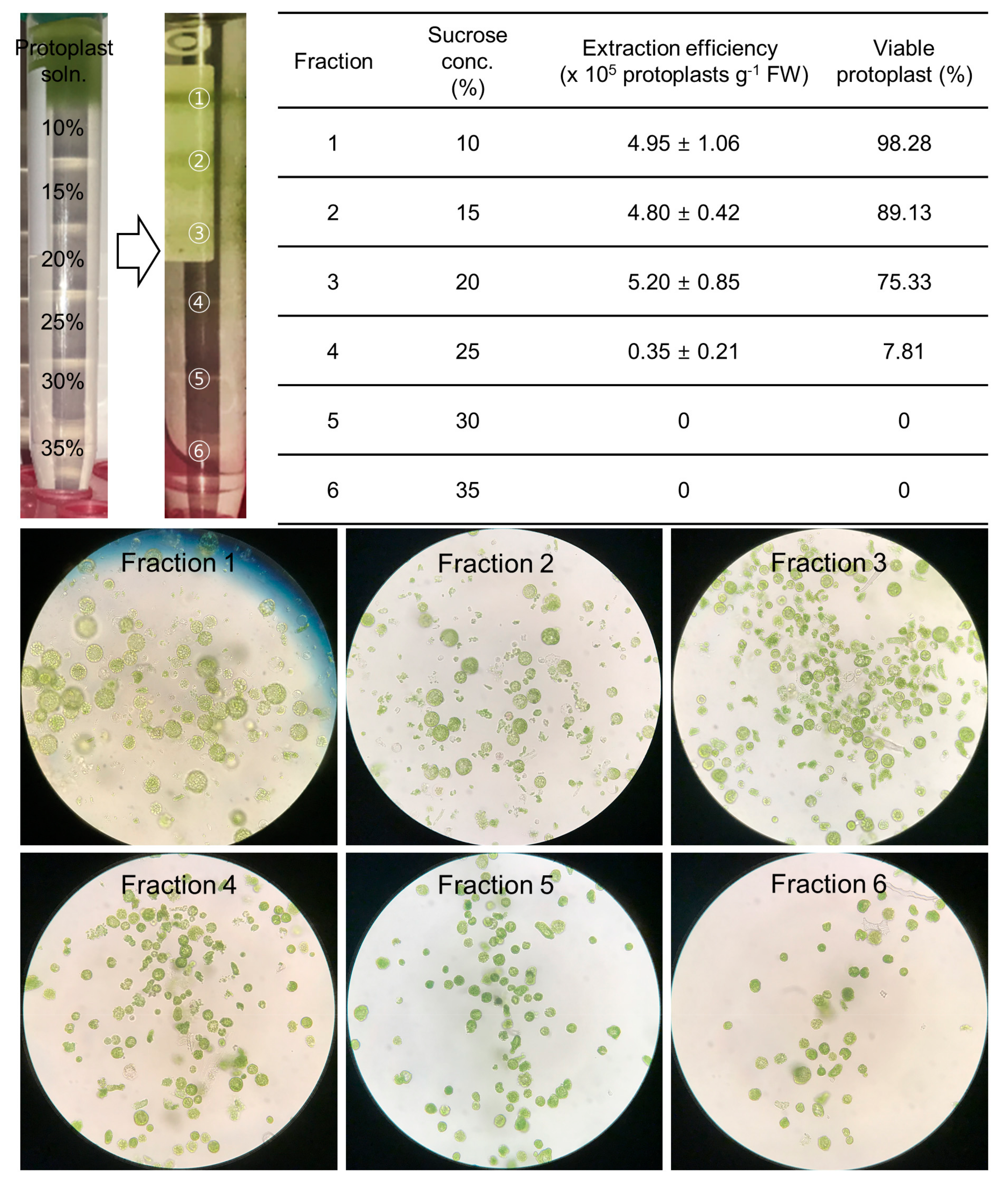
2.3. The Purification of High-Quality Protoplasts Was Accomplished by the Use of a Sucrose Gradient
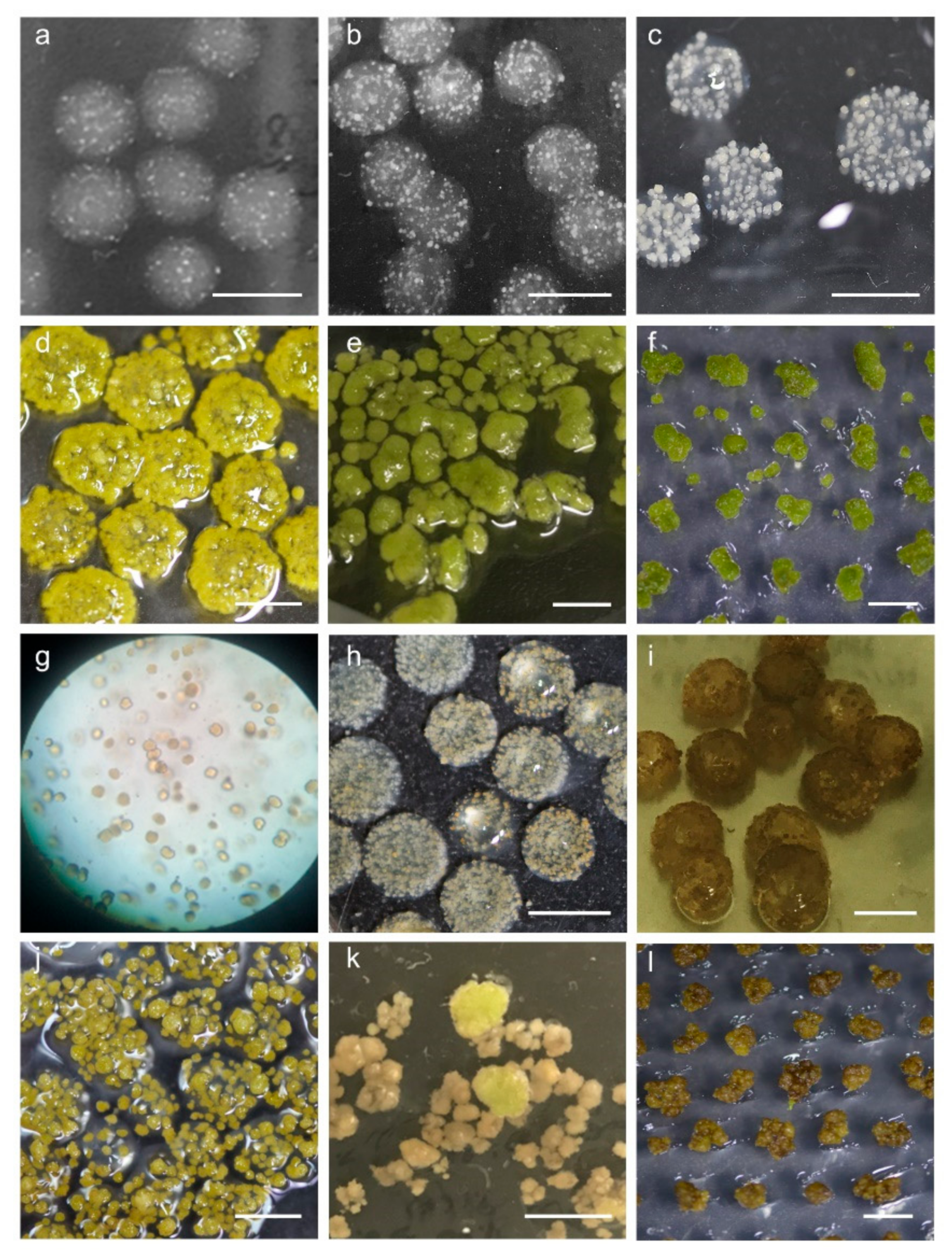
2.4. Establishment of Conditions for Microcallus Induction
2.5. Appropriate Space Is a Key Factor for Minicallus Development
2.6. Callus Greening Affects on Shoot Regeneration
3. Discussion
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. Preparation of Materials for Protoplasts
4.3. Protoplast Extraction and Purification
4.4. Protoplast Extraction from Enzymolysis Conditions
4.5. Protoplast Purification by Sucrose Gradient
4.6. Immobilization of Protoplasts in an Alginate Lens
4.7. Protoplast Culture and Plant Regeneration
4.8. Measurement of Regeneration Efficiency from Green Calli
4.9. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Upadhya, M.D. Isolation and culture of mesophyll protoplasts of potato (Solanum tuberosum L.). Potato Res. 1975, 18, 438–445. [Google Scholar] [CrossRef]
- Saravanan, S.; Sarvesan, R.; Vinod, M. Identification of DNA elements involved in somaclonal variants of rauvolfia serpentina (l.) arising from indirect organogenesis as evaluated by issr analysis. Indian J. Sci. Technol. 2011, 4, 1241–1245. [Google Scholar] [CrossRef]
- Sun, S.; Zhong, J.; Li, S.; Wang, X. Tissue culture-induced somaclonal variation of decreased pollen viability in torenia (torenia fournieri lind.). Bot. Stud. 2013, 54, 36. [Google Scholar] [CrossRef] [PubMed]
- Hare, K.; Dhurendra, S. Micropropagation of lasora (cordia myxa roxb.). Indian J. Hortic. 2013, 70, 323–327. [Google Scholar]
- Krishna, H.; Alizadeh, M.; Singh, D.; Singh, U.; Chauhan, N.; Eftekhari, M.; Sadh, R.K. Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech. 2016, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Arihara, A.; Kita, T.; Igarashi, S.; Goto, M.; Irikura, Y. White baron: A non-browning somaclonal variant of danshakuimo (irish cobbler). Am. Potato J. 1995, 72, 701–705. [Google Scholar] [CrossRef]
- Das, A.; Gosal, S.; Sidhu, J.; Dhaliwal, H. Induction of mutations for heat tolerance in potato by using in vitro culture and radiation. Euphytica 2000, 114, 205–209. [Google Scholar] [CrossRef]
- Veitia-Rodriguez, N.; Francisco-Cardoso, J.; Perez, J.; Garcia-Rodriguez, L.; Bermudez-Caraballosos, I.; Garcia-Rodriguez, L.; Padron-Montesinos, Y.; Orellana-Perez, P.; Romero-Quintana, C.; Hernandez, N. Evaluations in field of somaclones of irish potatoes (Solanum tuberosum L.) of the variety desiree obtained by somaclonal variation and in vitro mutagenesis. Biotechnol. Veg. 2002, 2, 21–26. [Google Scholar]
- Thieme, R.; Griess, H. Somaclonal variation in tuber traits of potato. Potato Res. 2005, 48, 153–165. [Google Scholar] [CrossRef]
- Nassar, A.M.; Kubow, S.; Leclerc, Y.N.; Donnelly, D.J. Somatic mining for phytonutrient improvement of ‘russet burbank’potato. Am. J. Potato Res. 2014, 91, 89–100. [Google Scholar] [CrossRef]
- Melchers, G.; Sacristán, M.D.; Holder, A.A. Somatic hybrid plants of potato and tomato regenerated from fused protoplasts. Carlsberg Res. Commun. 1978, 43, 203. [Google Scholar] [CrossRef]
- Sherraf, I.; Tizroutine, S.; Chaput, M.; Allot, M.; Mussio, I.; Sihachakr, D.; Rossignol, L.; Ducreux, G. Production and characterization of intergeneric somatic hybrids through protoplast electrofusion between potato (Solanum tuberosum) and lycopersicon pennellii. Plant. Cell Tisue Organ Cult. 1994, 37, 137–144. [Google Scholar] [CrossRef]
- Yu, Y.; Ye, W.; He, L.; Cai, X.; Liu, T.; Liu, J. Introgression of bacterial wilt resistance from eggplant to potato via protoplast fusion and genome components of the hybrids. Plant. Cell Rep. 2013, 32, 1687–1701. [Google Scholar] [CrossRef] [PubMed]
- Serraf, I.; Sihachakr, D.; Ducreux, G.; Brown, S.C.; Barghi, N.; Rossignol, L. Interspecific somatic hybridization in potato by protoplast electrofusion. Plant. Sci. 1991, 76, 115–126. [Google Scholar] [CrossRef]
- Cheng, J.; Saunders, J.A.; Sinden, S.L. Colorado potato beetle resistant somatic hybrid potato plants produced via protoplast electrofusion. In Vitro Cell. Dev. Biol. Plant 1995, 31, 90–95. [Google Scholar] [CrossRef]
- Jadari, R.; Sihachakr, D.; Rossignol, L.; Ducreux, G. Transfer of resistance to verticillium dahliae kleb. From solanum torvum sw into potato (Solanum tuberosum L.) by protoplast electrofusion. Euphytica 1992, 64, 39–47. [Google Scholar]
- Möllers, C.; Wenzel, G. Somatic hybridization of dihaploid potato protoplasts as a tool for potato breeding. Bot. Acta 1992, 105, 133–139. [Google Scholar] [CrossRef]
- Rasmussen, J.O.; Nepper, J.P.; Kirk, H.-G.; Tolstrup, K.; Rasmussen, O.S. Combination of resistance to potato late blight in foliage and tubers by intraspecific dihaploid protoplast fusion. Euphytica 1998, 102, 363–370. [Google Scholar] [CrossRef]
- Woestemeyer, A. In Transformation experiments with plasmid DNA [deoxyribonucleic acid] from agrobacterium tumefaciens in the potato [solanum tuberosum, germany, fr]. In Proceedings of the 23. Jahrestagung der Gesellschaft fuer Pflanzenbauwissenschaften eV Bonn, Bonn, Germany, 4–6 October 1979. [Google Scholar]
- Zhang, Y.; Liang, Z.; Zong, Y.; Wang, Y.; Liu, J.; Chen, K.; Qiu, J.-L.; Gao, C. Efficient and transgene-free genome editing in wheat through transient expression of crispr/cas9 DNA or rna. Nat. Commun. 2016, 7, 12617. [Google Scholar] [CrossRef] [PubMed]
- Malnoy, M.; Viola, R.; Jung, M.-H.; Koo, O.-J.; Kim, S.; Kim, J.-S.; Velasco, R.; Nagamangala Kanchiswamy, C. DNA-free genetically edited grapevine and apple protoplast using crispr/cas9 ribonucleoproteins. Front. Plant Sci. 2016, 7, 1904. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.W.; Kim, J.; Kwon, S.I.; Corvalán, C.; Cho, S.W.; Kim, H.; Kim, S.-G.; Kim, S.-T.; Choe, S.; Kim, J.-S. DNA-free genome editing in plants with preassembled crispr-cas9 ribonucleoproteins. Nat. Biotechnol. 2015, 33, 1162–1164. [Google Scholar] [CrossRef]
- Ali, M.; Mujib, A.; Zafar, N.; Tonk, D. Protoplast isolation and plant regeneration in two cultivated coriander varieties, co-1 and rs. Biotechnol. J. Biotechnol. Comput. Biol. Bionanotechnol. 2018, 99, 345–355. [Google Scholar] [CrossRef]
- Mukhtar, I.; Bajwa, R.; Nasim, G. Isolation of mesophyll protoplasts from leaves of dalbergia sissoo roxb. J. Appl. Sci. Environ. Manag. 2012, 16, 11–15. [Google Scholar]
- Andersson, M.; Turesson, H.; Olsson, N.; Fält, A.S.; Ohlsson, P.; Gonzalez, M.N.; Samuelsson, M.; Hofvander, P. Genome editing in potato via crispr-cas9 ribonucleoprotein delivery. Physiol. Plant. 2018, 164, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hunt, G.; Helgeson, J. A medium and simplified procedure for growing single cells from solanum species. Plant Sci. 1989, 60, 251–257. [Google Scholar] [CrossRef]
- Kim, H.-S.; Euym, J.-W.; Kim, M.-S.; Lee, B.-C.; Mook-Jung, I.; Jeon, J.-H.; Joung, H. Expression of human β-amyloid peptide in transgenic potato. Plant Sci. 2003, 165, 1445–1451. [Google Scholar] [CrossRef]
- Nicolia, A.; Proux-Wéra, E.; Åhman, I.; Onkokesung, N.; Andersson, M.; Andreasson, E.; Zhu, L.-H. Targeted gene mutation in tetraploid potato through transient talen expression in protoplasts. J. Biotechnol. 2015, 204, 17–24. [Google Scholar] [CrossRef]
- Tavazza, R.; Ancora, G. Plant regeneration from mesophyll protoplasts in commercial potato cultivars (primura, kennebec, spunta, desirée). Plant Cell Rep. 1986, 5, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Foulger, D.; Jones, M. Improved efficiency of genotype-dependent regeneration from protoplasts of important potato cultivars. Plant Cell Rep. 1986, 5, 72–76. [Google Scholar] [CrossRef]
- Chang, M.-M.; Loescher, W.H. Effects of preconditioning and isolation conditions on potato (Solanum tuberosum L. Cv. Russet burbank) protoplast yield for shoot regeneration and electroporation. Plant Sci. 1991, 73, 103–109. [Google Scholar] [CrossRef]
- El-Gioushy, S.F.E.E.-S.; Kareem, A.; Baiea, M.H.M. Pre-isolation, isolation and regeneration protoplasts from leaf mesophyll of in vivo malus domestica ‘anna’cv. Rev. Bras. Frutic. 2019, 41. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Cheng, J.; Zhao, W.; Li, X.; Wang, H.; Zhang, Z.; Sui, X. An efficient cucumber (Cucumis sativus L.) protoplast isolation and transient expression system. Sci. Hortic. 2013, 150, 206–212. [Google Scholar] [CrossRef]
- Mliki, A.; Jardak, R.; Reustle, G.M.; Ghorbel, A. Isolation and culture of leaf protoplasts from tunisian grapes. OENO One 2003, 37, 145–153. [Google Scholar] [CrossRef]
- Lee, N.; Wetzstein, H.Y. Protoplast isolation and callus production from leaves of tissue-cultured Vitis spp. Plant Cell Rep. 1988, 7, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Seo, P.J.; Cho, S.-H.; Park, C.-M. Preparation of leaf mesophyll protoplasts for transient gene expression in brachypodium distachyon. J. Plant Biol. 2012, 55, 390–397. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Zhan, J.; Huang, W.; Xu, H. An efficient peg-mediated transient gene expression system in grape protoplasts and its application in subcellular localization studies of flavonoids biosynthesis enzymes. Sci. Hortic. 2015, 191, 82–89. [Google Scholar] [CrossRef]
- Yao, L.; Liao, X.; Gan, Z.; Peng, X.; Wang, P.; Li, S.; Li, T. Protoplast isolation and development of a transient expression system for sweet cherry (Prunus avium L.). Sci. Hortic. 2016, 209, 14–21. [Google Scholar] [CrossRef]
- Tudses, N.; Premjet, S.; Premjet, D. Optimal conditions for high-yield protoplast isolations of Jatropha curcas L. and Ricinus communis L. Am. Eurasian J. Agric. Environ. Sci. 2014, 14, 221–230. [Google Scholar]
- Bai, L.; Cheng, Y.; She, J.; He, Z.; Liu, H.; Zhang, G.; Cao, R.; Chen, Y. Development of an efficient protoplast isolation and transfection system for castor bean (Ricinus communis L.). Plant Cell Tissue Organ Cult. 2020, 143, 457–464. [Google Scholar] [CrossRef]
- Davey, M.R.; Anthony, P.; Power, J.B.; Lowe, K.C. Plant protoplast technology: Current status. Acta Physiol. Plant. 2005, 27, 117–130. [Google Scholar] [CrossRef]
- Hughes, B.G.; White, F.G.; Smith, M.A. Purification of plant protoplasts by discontinuous gradient centrifugation. Biochem. Physiol. Pflanz. 1978, 172, 223–231. [Google Scholar] [CrossRef]
- Perales, E.H.; Schieder, O. Plant regeneration from leaf protoplasts of apple. Plant Cell Tissue Organ Cult. 1993, 34, 71–76. [Google Scholar] [CrossRef]
- Schilde-Rentschler, L.; Boos, G.; Ninnemann, H. Somatic hybridization of diploid potato lines, a tool in potato breeding. In Progress in Plant Protoplast Research; Springer: Dordrecht, The Netherlands, 1988; pp. 195–196. [Google Scholar]
- Damm, B.; Willmitzer, L. Regeneration of fertile plants from protoplasts of different arabidopsis thaliana genotypes. Mol. Gen. Genet. 1988, 213, 15–20. [Google Scholar] [CrossRef]
- Grzebelus, E.; Szklarczyk, M.; Baranski, R. An improved protocol for plant regeneration from leaf-and hypocotyl-derived protoplasts of carrot. Plant Cell Tissue Organ Cult. 2012, 109, 101–109. [Google Scholar] [CrossRef]
- Ehsanpour, A.; Jones, M. Plant regeneration from mesophyll protoplasts of potato (solanum tuberosum l.) cultivar delaware using silver thiosulfate (sts). J. Sci. I. R. Iran 2001, 12, 103–110. [Google Scholar]
- Andersson, M.; Turesson, H.; Nicolia, A.; Fält, A.-S.; Samuelsson, M.; Hofvander, P. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient crispr-cas9 expression in protoplasts. Plant Cell Rep. 2017, 36, 117–128. [Google Scholar] [CrossRef]
- Sadia, B. Improved isolation and culture of protoplasts from S. chacoense and potato: Morphological and cytological evaluation of protoplast-derived regenerants of potato cv. Desiree. Pak. J. Agric. Sci. 2015, 52, 51–61. [Google Scholar]
- Cui, J.; Mackenzie, K.K.; Eeckhaut, T.; Müller, R.; Lütken, H. Protoplast isolation and culture from kalanchoë species: Optimization of plant growth regulator concentration for efficient callus production. Plant Cell Tissue Organ Cult. 2019, 138, 287–297. [Google Scholar] [CrossRef]
- Guri, A.; Volokita, M.; Sink, K. Plant regeneration from leaf protoplasts of solanum torvum. Plant Cell Rep. 1987, 6, 302–304. [Google Scholar] [CrossRef]
- Borgato, L.; Pisani, F.; Furini, A. Plant regeneration from leaf protoplasts of solanum virginianum l.(solanaceae). Plant Cell Tissue Organ Cult. 2007, 88, 247–252. [Google Scholar] [CrossRef]
- Rahmani, M.-S.; Pijut, P.M.; Shabanian, N. Protoplast isolation and genetically true-to-type plant regeneration from leaf-and callus-derived protoplasts of albizia julibrissin. Plant Cell Tissue Organ Cult. 2016, 127, 475–488. [Google Scholar] [CrossRef]
- Hassanein, A.; Dorion, N. High-efficiency colony formation and whole plant regeneration from mesophyll protoplasts of pelargonium× hortorum ‘panaché sud’. J. Hortic. Sci. Biotechnol. 2006, 81, 714–720. [Google Scholar] [CrossRef]
- Gauchan, D. Effect of different sugars on shoot regeneration of maize (zea mays L.). Kathmandu Univ. J. Sci. Eng. Technol. 2012, 8, 119–124. [Google Scholar] [CrossRef]
- van Rensburg, J.G.; Vcelar, B.M. The effect of the sucrose concentration on the initiation and growth of adventitious buds from leaf tissue of lachenalia. South Afr. J. Bot. 1989, 55, 117–121. [Google Scholar] [CrossRef]
- Godo, T.; Matsui, K.; Kida, T.; Mii, M. Effect of sugar type on the efficiency of plant regeneration from protoplasts isolated from shoot tip-derived meristematic nodular cell clumps of lilium x formolongi hort. Plant Cell Rep. 1996, 15, 401–404. [Google Scholar] [CrossRef]
- Shepard, J.F.; Totten, R.E. Mesophyll cell protoplasts of potato: Isolation, proliferation, and plant regeneration. Plant Physiol. 1977, 60, 313–316. [Google Scholar] [CrossRef]
- Fossi, M.; Amundson, K.; Kuppu, S.; Britt, A.; Comai, L. Regeneration of solanum tuberosum plants from protoplasts induces widespread genome instability. Plant Physiol. 2019, 180, 78–86. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Park, S.; Jeon, J.H.; Kim, H.S.; Park, Y.; Aswath, C.; Joung, H. Effect of sealed and vented gaseous microenvironments on the hyperhydricity of potato shoots in vitro. Sci. Hortic. 2004, 99, 199–205. [Google Scholar] [CrossRef]
- Moon, K.-B.; Ahn, D.-J.; Park, J.-S.; Jung, W.Y.; Cho, H.S.; Kim, H.-R.; Jeon, J.-H.; Park, Y.-i.; Kim, H.-S. Transcriptome profiling and characterization of drought-tolerant potato plant (Solanum tuberosum L.). Mol. Cells 2018, 41, 979. [Google Scholar]
- Wu, F.-H.; Shen, S.-C.; Lee, L.-Y.; Lee, S.-H.; Chan, M.-T.; Lin, C.-S. Tape-arabidopsis sandwich-a simpler arabidopsis protoplast isolation method. Plant Methods 2009, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]






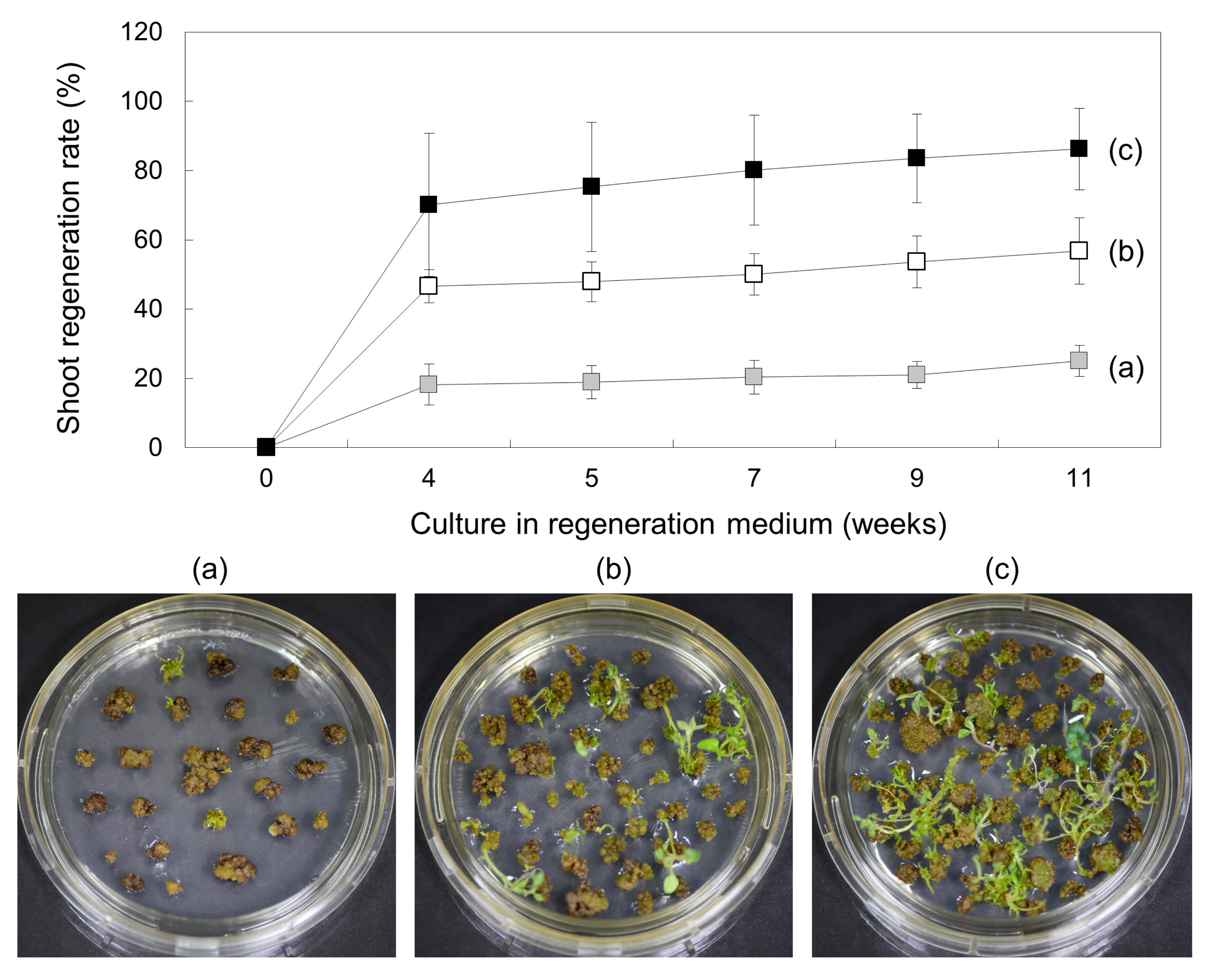
 , (a)), 5 weeks (
, (a)), 5 weeks ( , (b)), and 6 weeks (
, (b)), and 6 weeks ( , (c)) and then transferred to SI2 medium for shoot induction. Regeneration efficiency was measured using an average of 60 calli clumps per replicate. Three to five replicates were averaged. Error bars indicate ± standard deviation of the mean (SD).
, (a)), 5 weeks (, (b)), and 6 weeks (, (c)) and then transferred to SI2 medium for shoot induction. Regeneration efficiency was measured using an average of 60 calli clumps per replicate. Three to five replicates were averaged. Error bars indicate ± standard deviation of the mean (SD).
, (c)) and then transferred to SI2 medium for shoot induction. Regeneration efficiency was measured using an average of 60 calli clumps per replicate. Three to five replicates were averaged. Error bars indicate ± standard deviation of the mean (SD).
, (a)), 5 weeks (, (b)), and 6 weeks (, (c)) and then transferred to SI2 medium for shoot induction. Regeneration efficiency was measured using an average of 60 calli clumps per replicate. Three to five replicates were averaged. Error bars indicate ± standard deviation of the mean (SD).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Wound Type | Extraction Efficiency (×105 Protoplasts/g FW) | p-Value (Student’s t-Test) | ||
|---|---|---|---|---|---|
| vs. W1 | vs. W2 | vs. W3 | |||
| W1 | Peeling off | 4.45 ± 1.23 | - | 0.1820 | 0.0194 |
| W2 | Gentle chopping | 7.14 ± 2.38 | 0.1820 | - | 0.0473 |
| W3 | Leaf strips | 13.23 ± 2.80 | 0.0194 | 0.0473 | - |
| Enzymolysis Time | Enzyme Concentration | Wound Type | Extraction Efficiency (×105 protoplasts/g FW) | p-Value (Student’s t-Test) | |||
|---|---|---|---|---|---|---|---|
| Cellulase R-10 (%, w/v) | Macerozyme (%, w/v) | vs. 5 h | vs. 18 h | vs. 24 h | |||
| 5 h | 1 | 0.2 | Leaf strips | 5.24 ± 1.90 | - | 0.0165 | 0.0492 |
| 18 h | 1 | 0.2 | Leaf strips | 14.20 ± 2.96 | 0.0165 | - | 0.0942 |
| 24 h | 1 | 0.2 | Leaf strips | 9.38 ± 0.88 | 0.0492 | 0.0942 | - |
| Conditions | Medium | Cellulase/ Macerozyme (%, w/v) | Volume of Wash Soln. | Shaking (rpm) | Plasmolysis Time (h) | Enzymolysis Time (h) | Extraction Efficiency (×105 Protoplasts/g FW) |
|---|---|---|---|---|---|---|---|
| M1 | 20 mM 2-(N-Morpholino)ethanesulfonic Acid (MES) 0.5 M mannitol 20 mM KCl 10 mM CaCl2 0.1% Bovine Serum Albumin (BSA) pH 5.7 | 1/0.2 | 1× | 0 | 1 | 5 | 5.24 ± 1.90 |
| 6 | 8.56 ± 1.84 | ||||||
| 18 | 12.27 ± 3.40 | ||||||
| M2 | 20 mM MES 0.5 M mannitol 20 mM KCl 10 mM CaCl2 0.1% BSA pH 5.7 | 1/0.5 | 1× | 40 | 0 | 5 | 21.09 ± 8.49 |
| 6 | 39.63 ± 4.09 | ||||||
| 18 | 28.14 ± 3.34 | ||||||
| M3 | 20 mM MES 0.5 M mannitol 20 mM KCl 10 mM CaCl2 0.1% BSA pH 5.7 | 1/0.5 | 3× | 40 | 0 | 5 | 36.40 ± 4.66 |
| 6 | 63.59 ± 5.14 | ||||||
| 18 | N/A |
| Abbreviation | Basal Medium | Components | Purpose |
|---|---|---|---|
| E1 | 20 mM MES, 1% cellulase ‘Onozuka R10′, 0.5% macerozyme, 0.5 M mannitol, 20 mM KCl, 10 mM CaCl2, 0.1% BSA, pH 5.8 (autoclaved) | Protoplast extraction | |
| W | Macro C and micro C stocks, iron elements C, 2.0 mg/L α-Naphthaleneacetic Acid (NAA), 0.5 mg/L 6-Benzylaminopurine (BAP), 14.03 g/L NaCl, pH 5.6 | Protoplast washing | |
| CI1 | Macro B and micro B stocks, iron elements B, carbohydrates B, vitamins B, 1.0 mg/L NAA, 0.4 mg/L BAP, and other organics, pH 5.6 | Microcallus induction | |
| CI2 | Macro A and micro A stocks, iron elements A, carbohydrate A, vitamins A, 1 g/L BSA, 1.0 mg/L NAA, 0.4 mg/L BAP, and other organics, pH 5.6 | Microcallus induction | |
| P | MS | 107 mg/L NH4Cl, 2.5 g/L sucrose, 54.7 g/L mannitol, 40 mg/L adenine sulfate, 0.1 g/L casein hydrolysate, vitamins C, 0.1 mg/L NAA, 0.5 mg/L BAP | Proliferation |
| G | MS | 267.5 mg/L NH4Cl, 2.5 g/L sucrose, 36.4 g/L mannitol, 80 mg/L adenine sulfate, 0.1 g/L casein hydrolysate, vitamins C, 0.1 mg/L IAA, 2.5 mg/L zeatin | Callus greening |
| SI1 | MS | 30 g/L sucrose, 0.01 mg/L NAA, 2.0 mg/L zeatin, 0.1 mg/L Gibberellic acid (GA3), 4.0 g/L plant agar, pH 5.6 | Shoot regeneration |
| SI2 | MS | 10 g/L sucrose, no vitamins, no myo-inositol, 0.01 mg/L NAA, 2.0 mg/L zeatin, 0.1 mg/L GA3, 2.5 g/L gelrite, pH 5.6 | Shoot regeneration |
| Microcallus Medium | Protoplasts/ Alginate Lens (A) | Callus Counts/ Alginate Lens (B) | Callus Induction Efficiency (%) (B/A × 100) |
|---|---|---|---|
| CI1 | 2 × 103 | 125.13 ± 15.01 | 6.26 ± 0.75 |
| CI2 | 2 × 103 | 147.95 ± 10.06 | 7.40 ± 0.50 |
| Macro-Elements | A (mg/L) | B (mg/L) | C (mg/L) |
| KNO3 | 740 | 1900 | 740 |
| MgSO4·7H2O | 492 | 350 | 492 |
| KH2PO4 | 34 | 680 | 34 |
| CaCl2·2H2O | 368 | 600 | 882 |
| Iron Elements | A | B | C |
| Na2EDTA | 14 | 37.3 | 14 |
| FeSO4·7H2O | 19 | 27.8 | 19 |
| Micro-Elements | A | B | C |
| H3BO3 | 1.5 | 3 | 1.5 |
| MnSO4·H2O | 5 | 10 | 5 |
| ZnSO4·7H2O | 1 | 2 | 1 |
| Na2MoO4·2H2O | 0.12 | 0.25 | 0.12 |
| CuSO4·5H2O | 0.012 | 0.025 | 0.012 |
| CoCl2·6H2O | 0.012 | 0.025 | 0.012 |
| KI | 0.38 | 0.75 | 0.38 |
| Carbohydrates (g/L) | A | B | |
| Glucose | 33.7 | 10 | |
| Mannitol | 30.92 | 40 | |
| Sucrose | 0.125 | 10 | |
| Sorbitol | 0.125 | 0.25 | |
| D(−)Fructose | 0.125 | 0.25 | |
| D(−)Ribose | 0.125 | 0.25 | |
| D(+)Xylose | 0.125 | 0.25 | |
| D(+)Mannose | 0.125 | 0.25 | |
| L(+)Rhamnose | 0.125 | 0.25 | |
| D(+)Cellobiose | 0.125 | 0.25 | |
| Myo-inositol | 0.05 | 0.05 | |
| Vitamins | A | B | C |
| Pantothenic acid | 2.5 | 0.5 | - |
| Choline Chloride | 2.5 | 0.5 | - |
| Ascorbic acid | 5 | 1 | - |
| p-Aminobenzoic acid | 0.05 | 0.01 | - |
| Nicotinic acid | 2.5 | 5 | 5 |
| Pyridoxine-HCl | 2.5 | 0.5 | 0.5 |
| Thiamine-HCl | 25 | 2 | 0.5 |
| Folic acid | 1 | - | 0.5 |
| Riboflavin | - | 0.1 | - |
| Biotin | 0.025 | 0.005 | 0.05 |
| Cyanocobalamin | 0.05 | - | - |
| Cholecalciferol (Vit. D) | 0.025 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, K.-B.; Park, J.-S.; Park, S.-J.; Lee, H.-J.; Cho, H.-S.; Min, S.-R.; Park, Y.-I.; Jeon, J.-H.; Kim, H.-S. A More Accessible, Time-Saving, and Efficient Method for In Vitro Plant Regeneration from Potato Protoplasts. Plants 2021, 10, 781. https://doi.org/10.3390/plants10040781
Moon K-B, Park J-S, Park S-J, Lee H-J, Cho H-S, Min S-R, Park Y-I, Jeon J-H, Kim H-S. A More Accessible, Time-Saving, and Efficient Method for In Vitro Plant Regeneration from Potato Protoplasts. Plants. 2021; 10(4):781. https://doi.org/10.3390/plants10040781
Chicago/Turabian StyleMoon, Ki-Beom, Ji-Sun Park, Su-Jin Park, Hyo-Jun Lee, Hye-Sun Cho, Sung-Ran Min, Youn-Il Park, Jae-Heung Jeon, and Hyun-Soon Kim. 2021. "A More Accessible, Time-Saving, and Efficient Method for In Vitro Plant Regeneration from Potato Protoplasts" Plants 10, no. 4: 781. https://doi.org/10.3390/plants10040781
APA StyleMoon, K.-B., Park, J.-S., Park, S.-J., Lee, H.-J., Cho, H.-S., Min, S.-R., Park, Y.-I., Jeon, J.-H., & Kim, H.-S. (2021). A More Accessible, Time-Saving, and Efficient Method for In Vitro Plant Regeneration from Potato Protoplasts. Plants, 10(4), 781. https://doi.org/10.3390/plants10040781