
Biomimetic Approaches to the Synthesis of Natural Disesquiterpenoids: An Update




Abstract
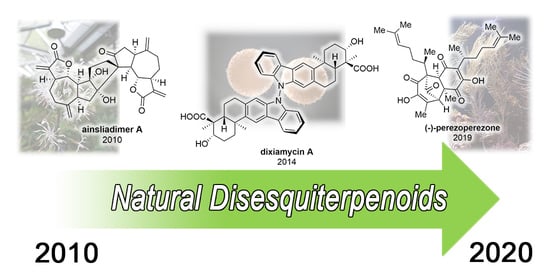
1. Introduction
2. Bisabolane Dimers
2.1. Meiogynin A
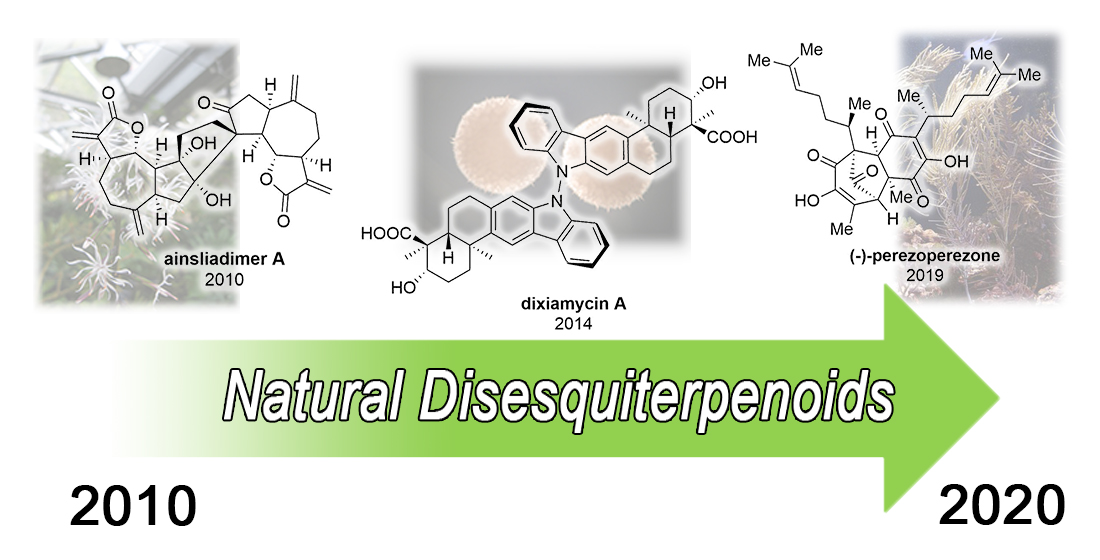
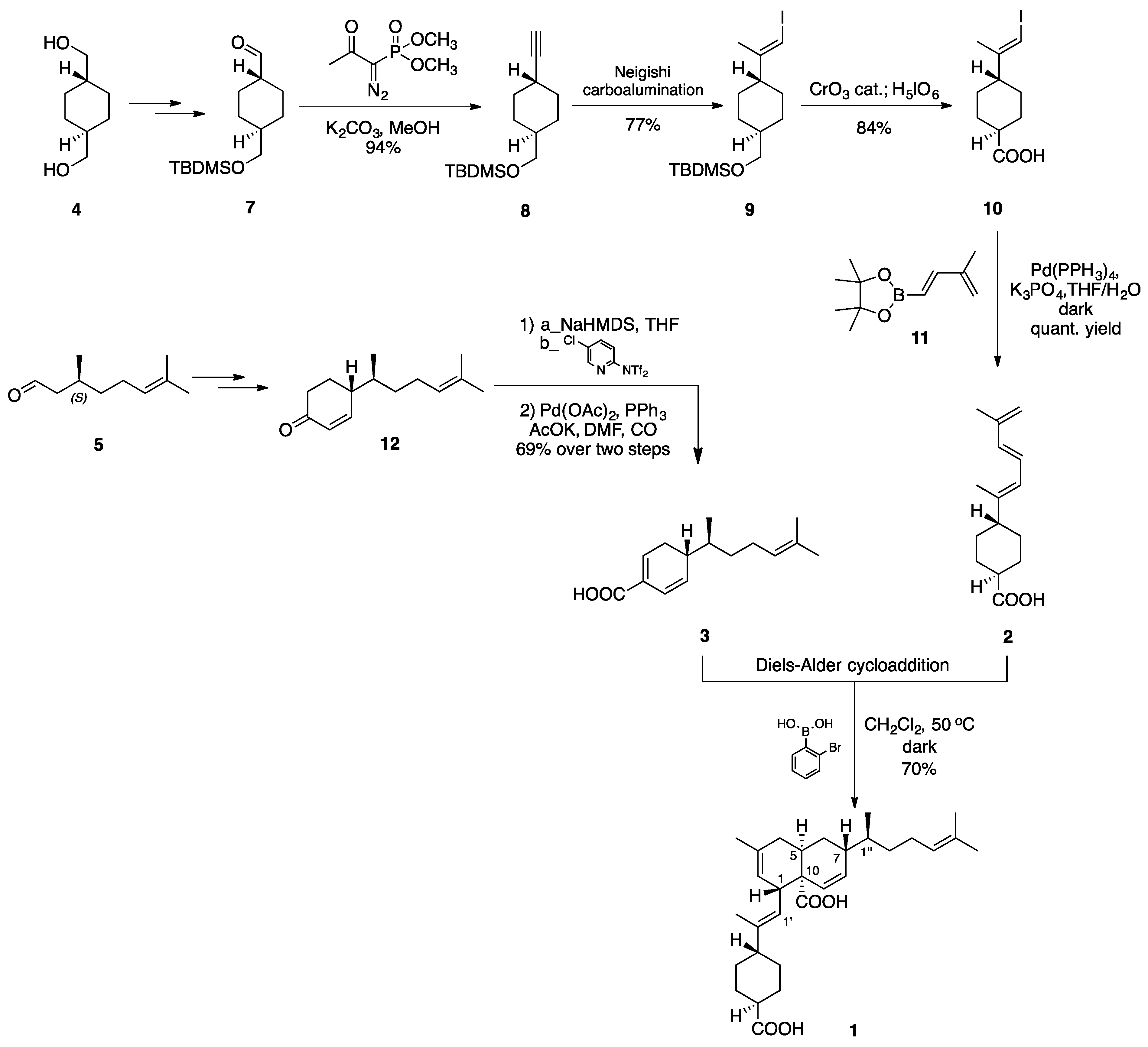
2.2. (–)-Perezoperezone
3. Xanthane Dimers
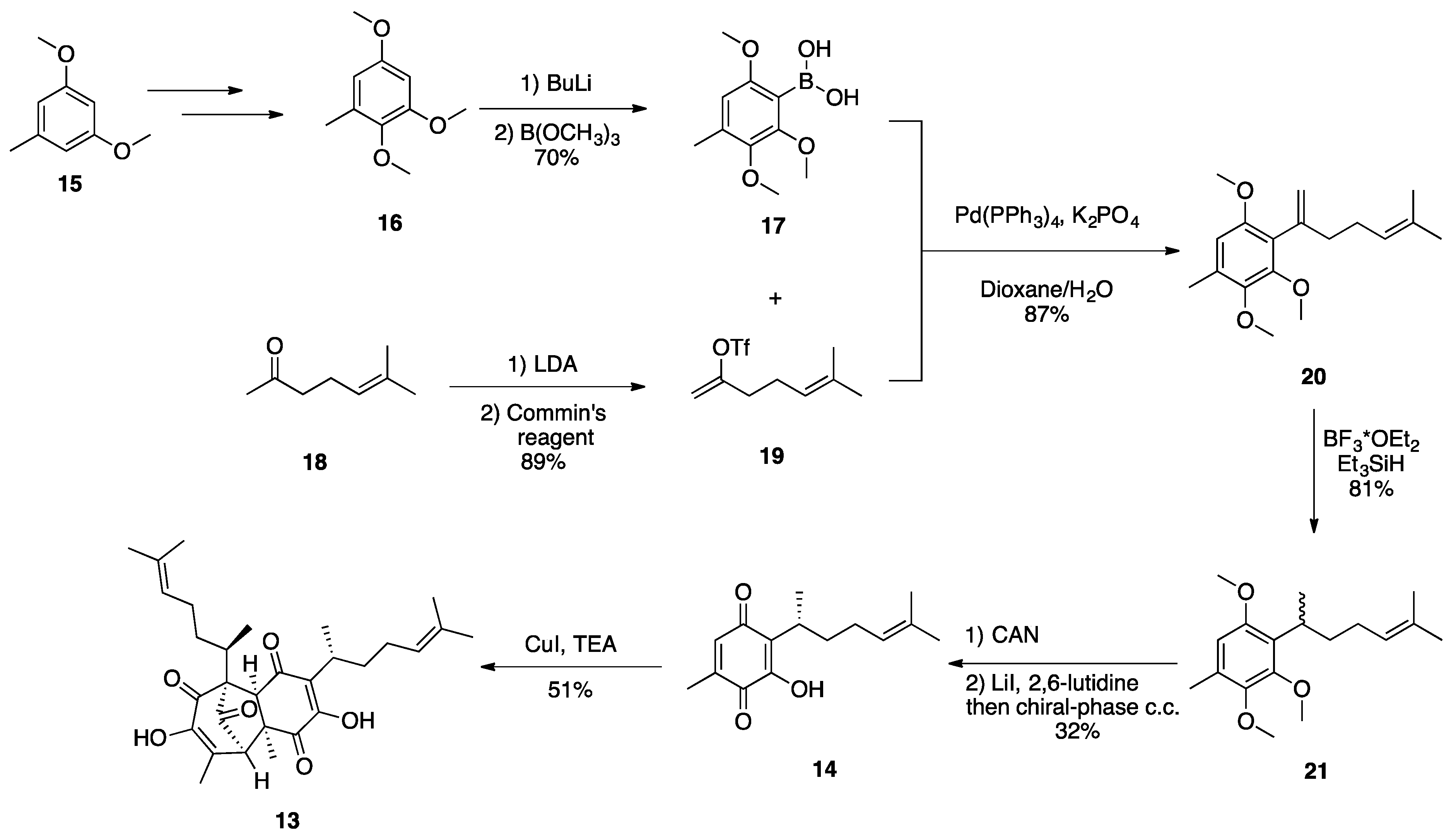
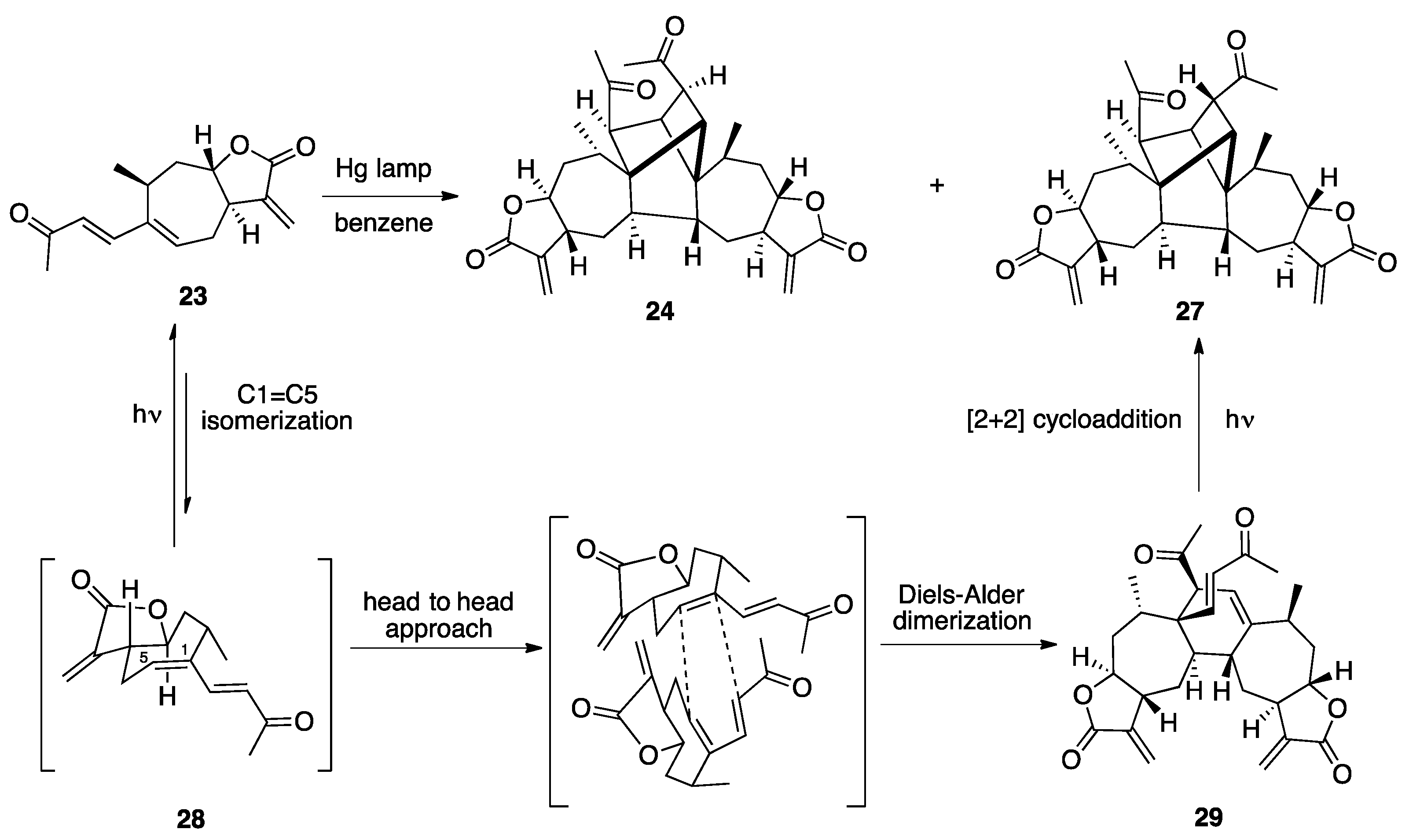
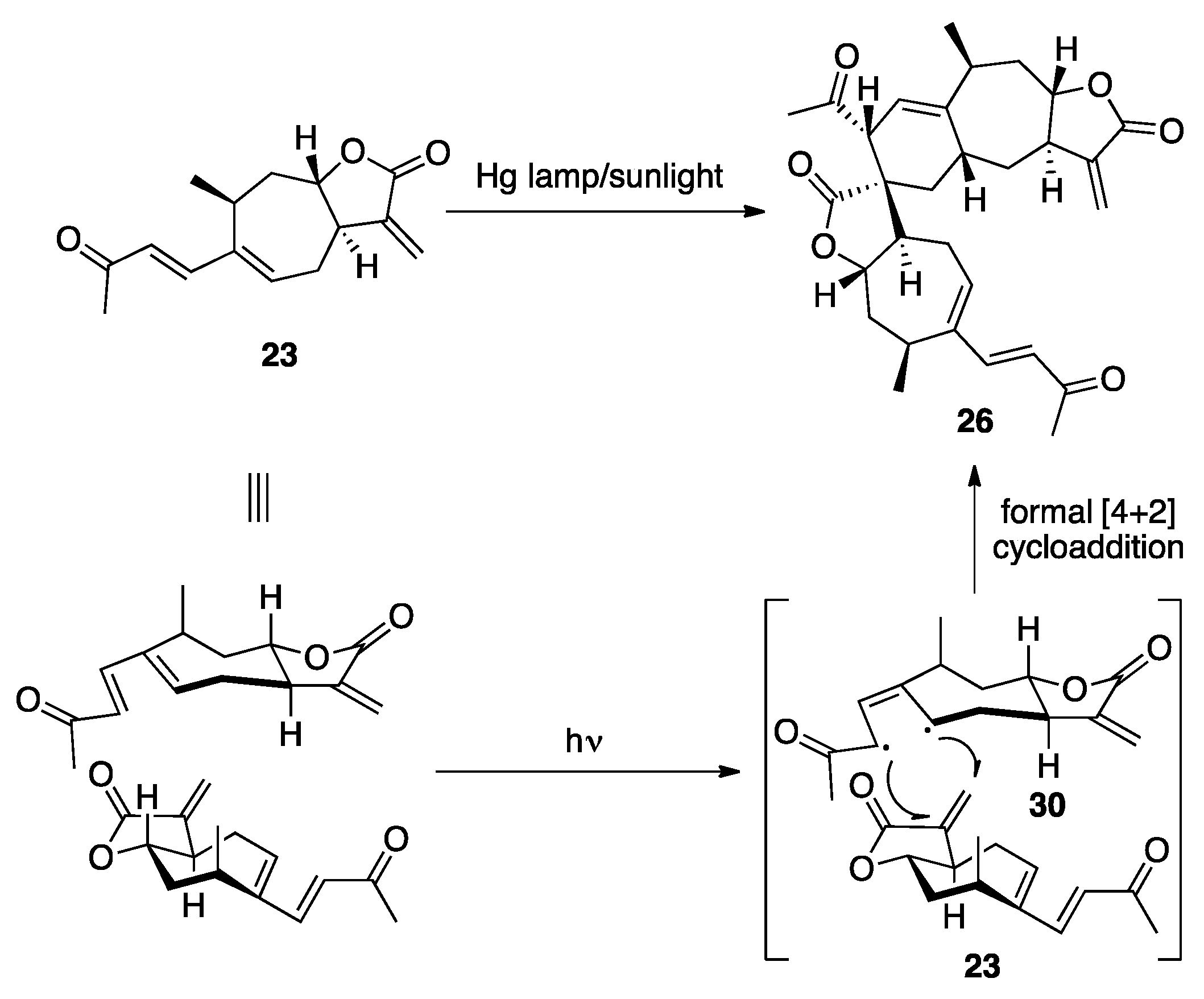
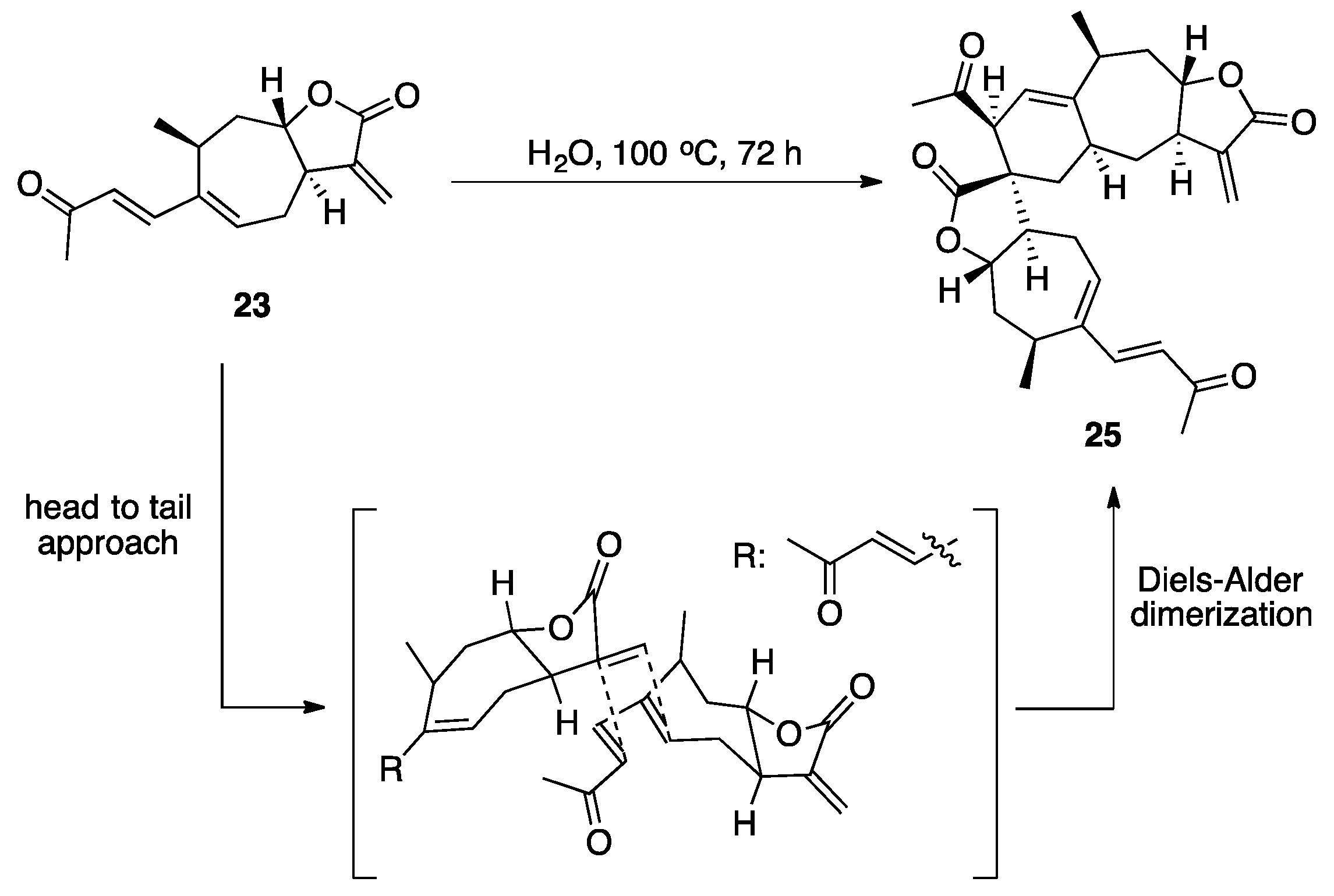
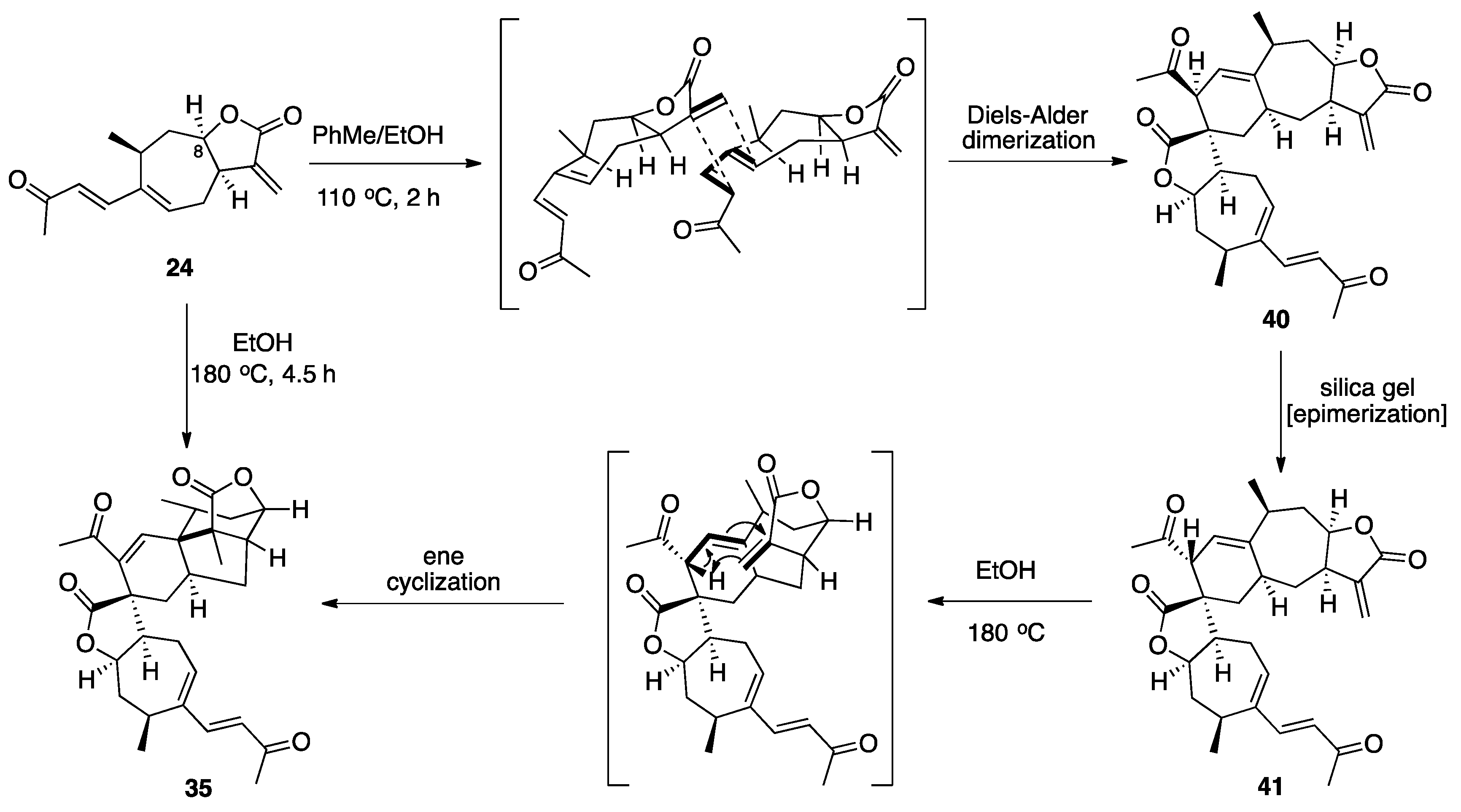
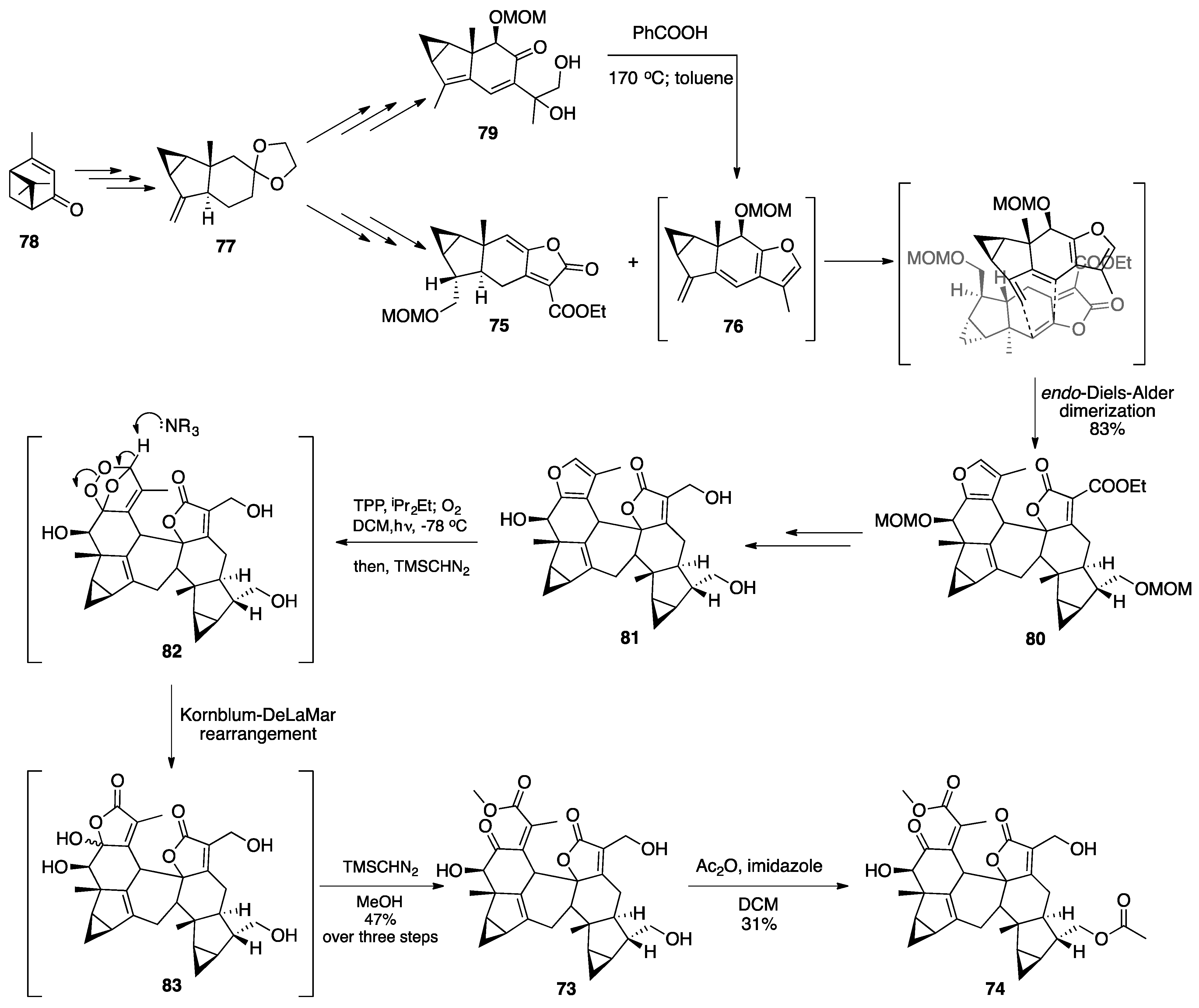
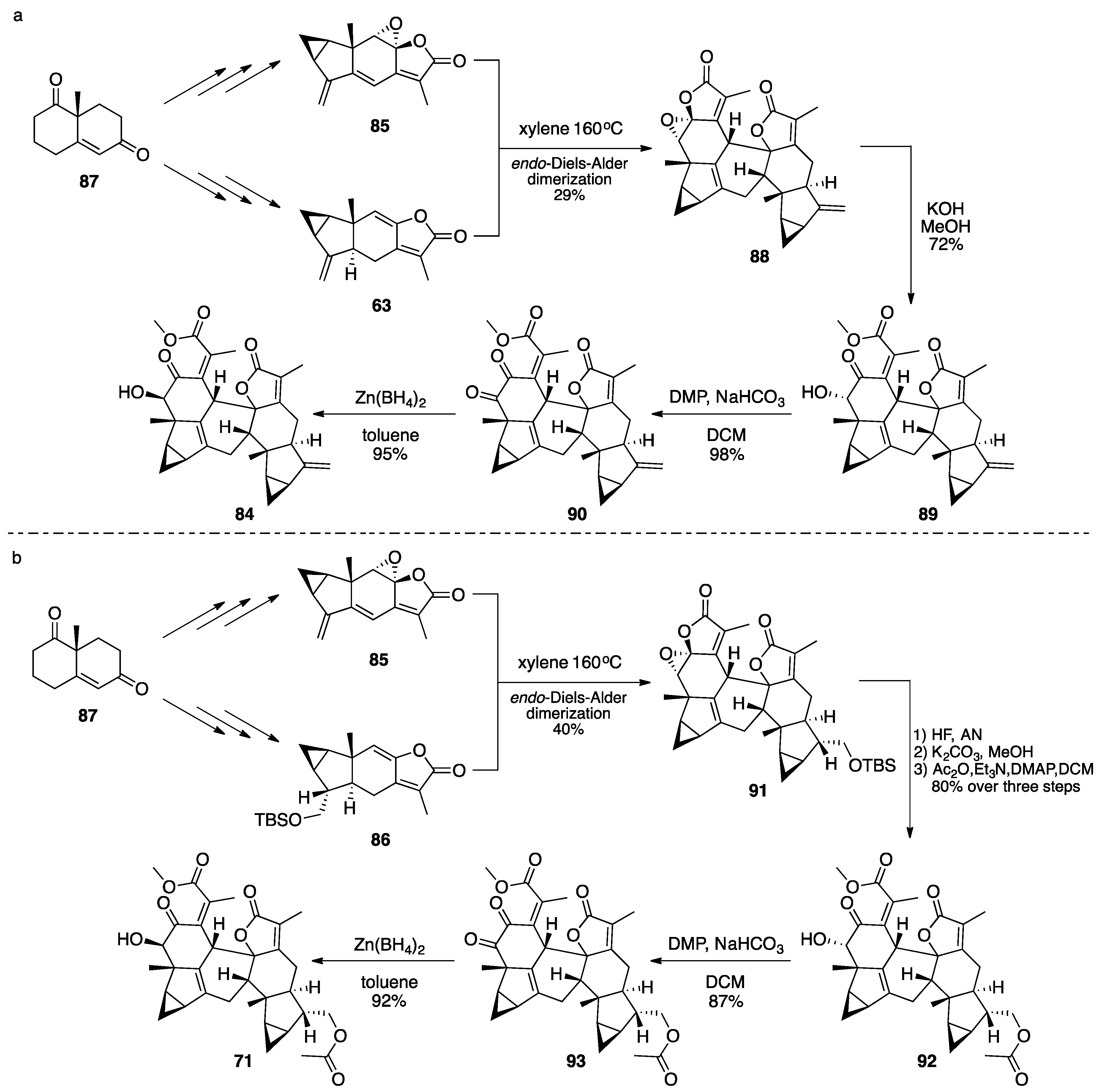
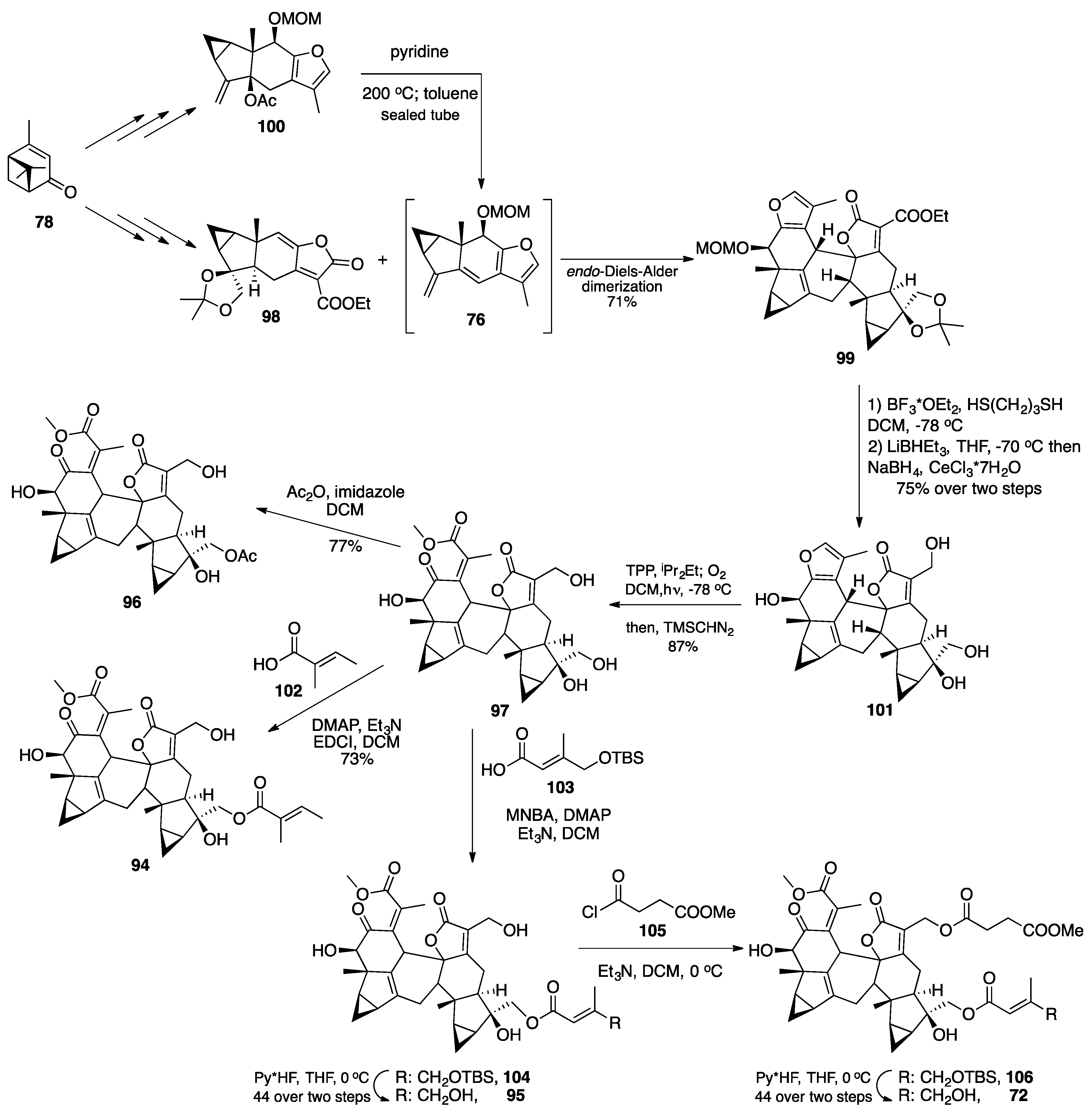
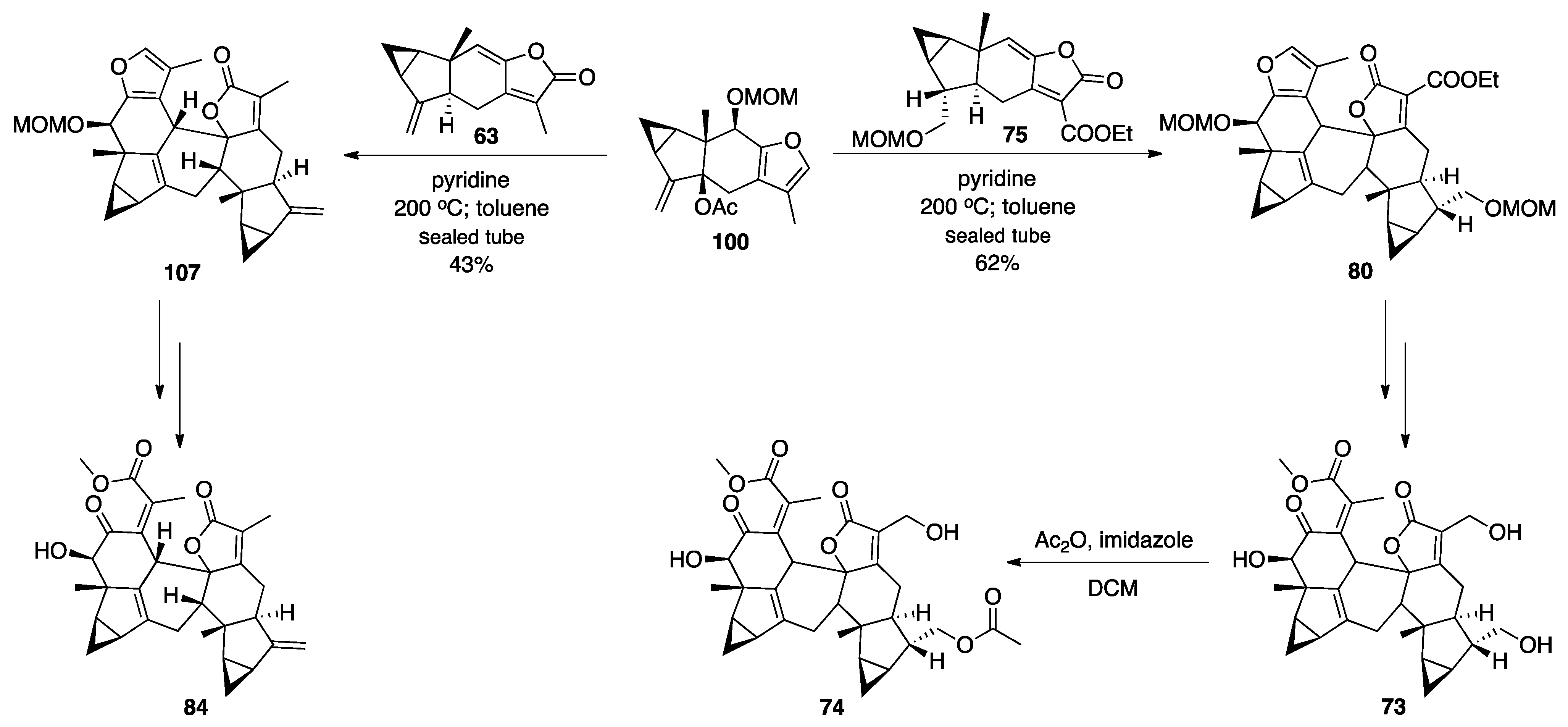
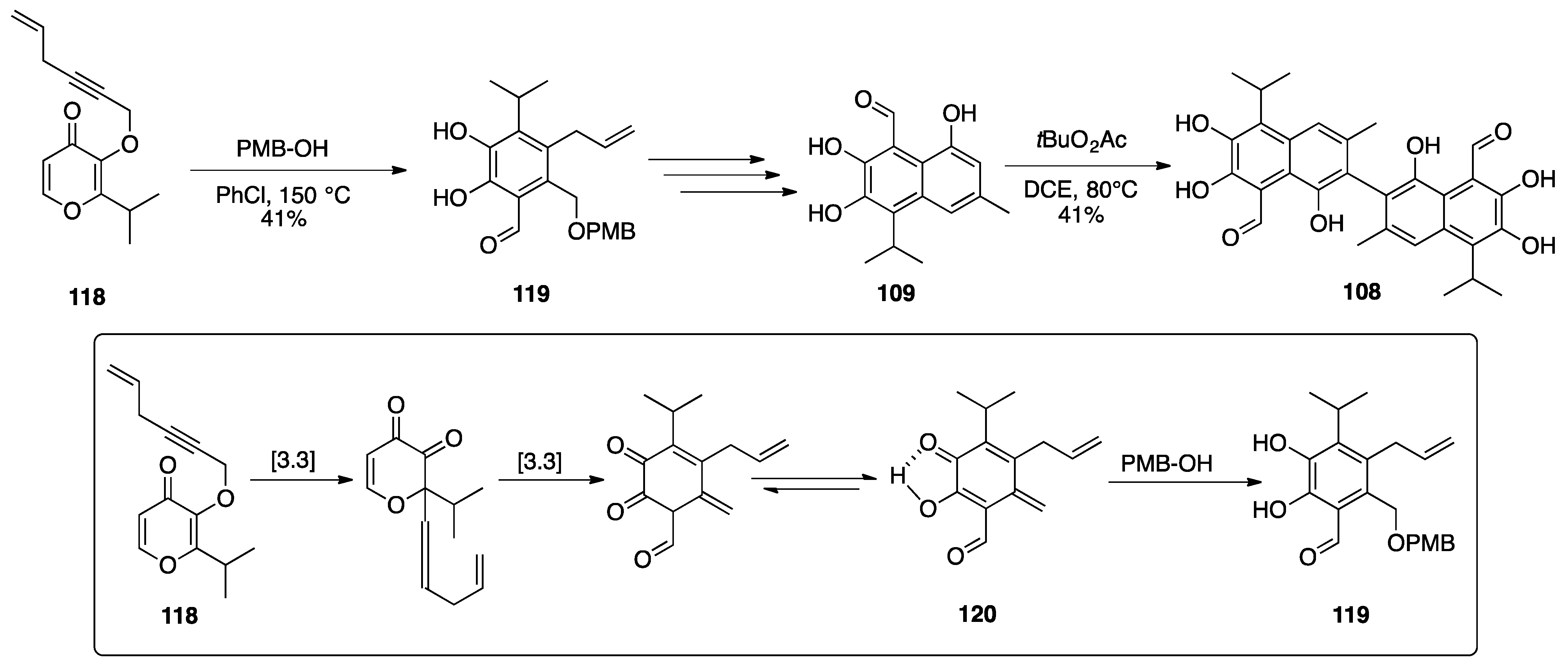
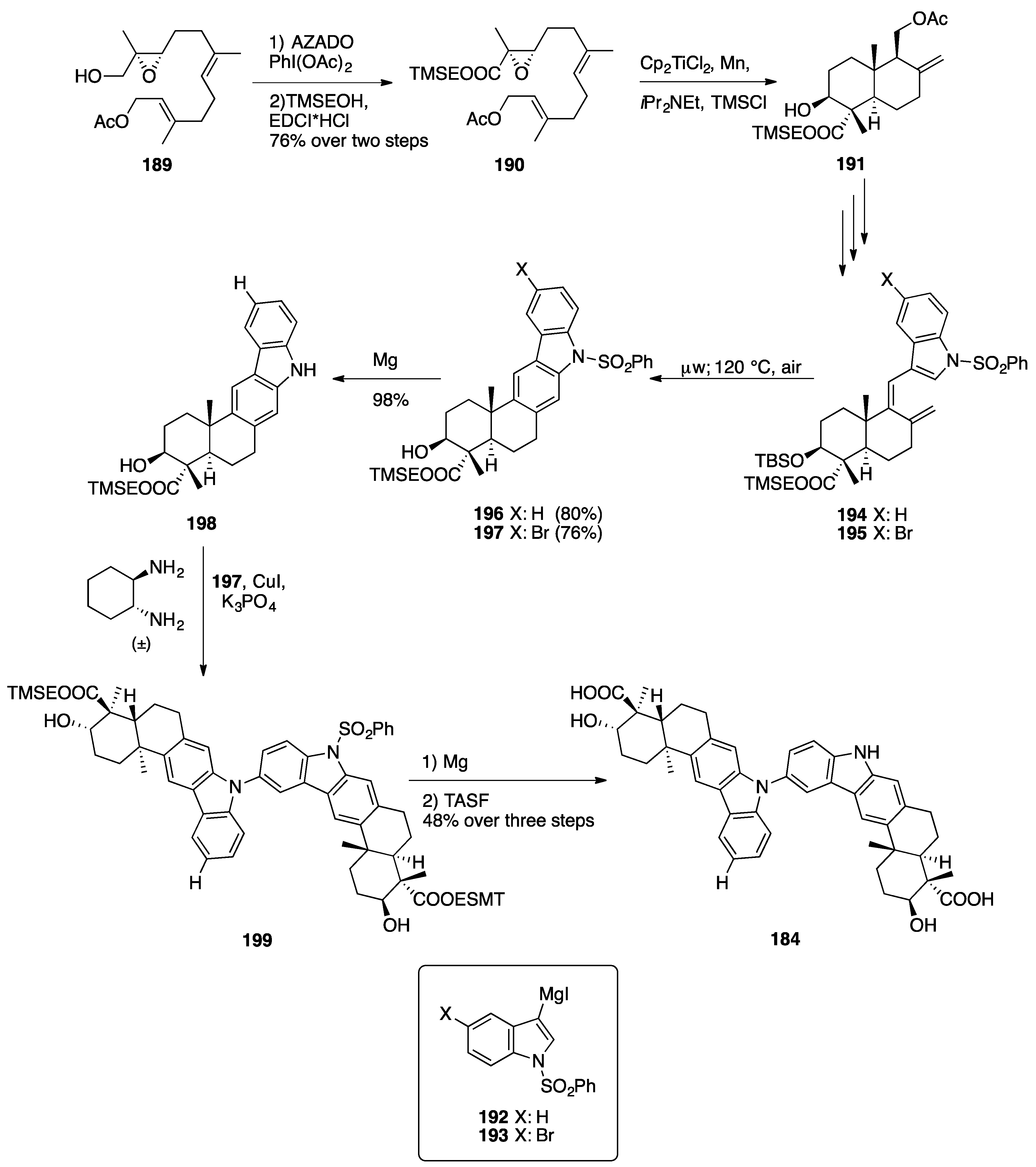
3.1. Mogolides A-C
3.2. Pungiolides A-E, L-N
4. Guaiane Dimers
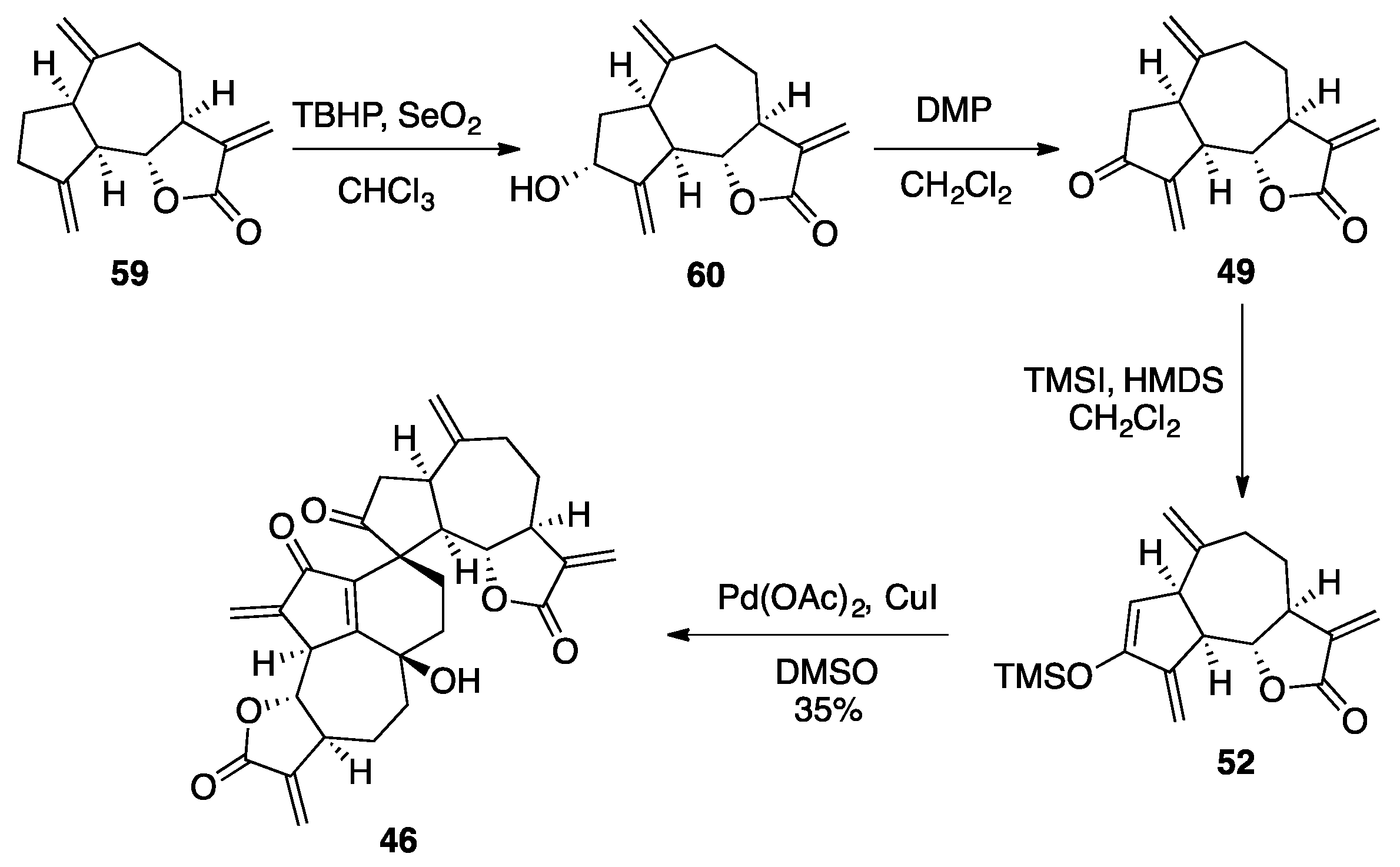
Ainsliadimer A, B and Gochnatiolides A-C
5. Lindenane Dimers
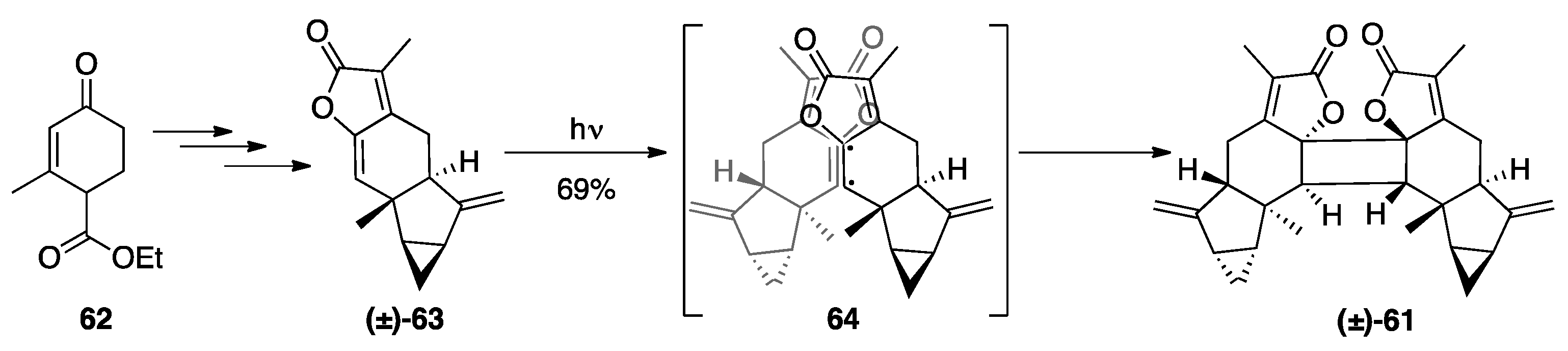
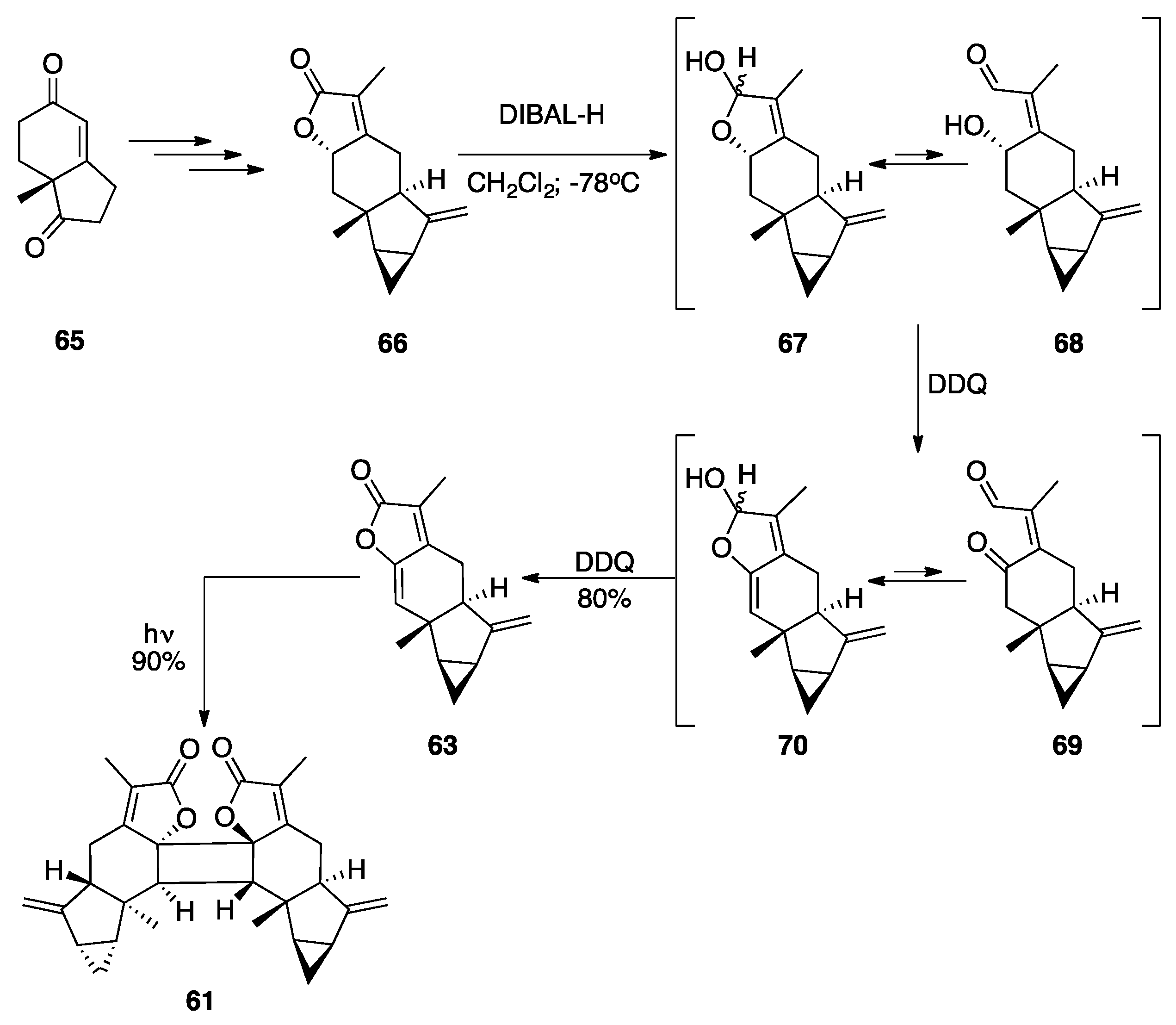
5.1. Lindenane [2+2] Dimers: Chloranthalactone F
5.2. Lindenane [4+2] Dimers: Shizukaols A, C, D, E, I, Chlorajaponilide C, Multistalide B, Sarcandrolide J and Sarglabolide I
6. Cadinane Dimers
Gossypol
7. Miscellaneous Dimers
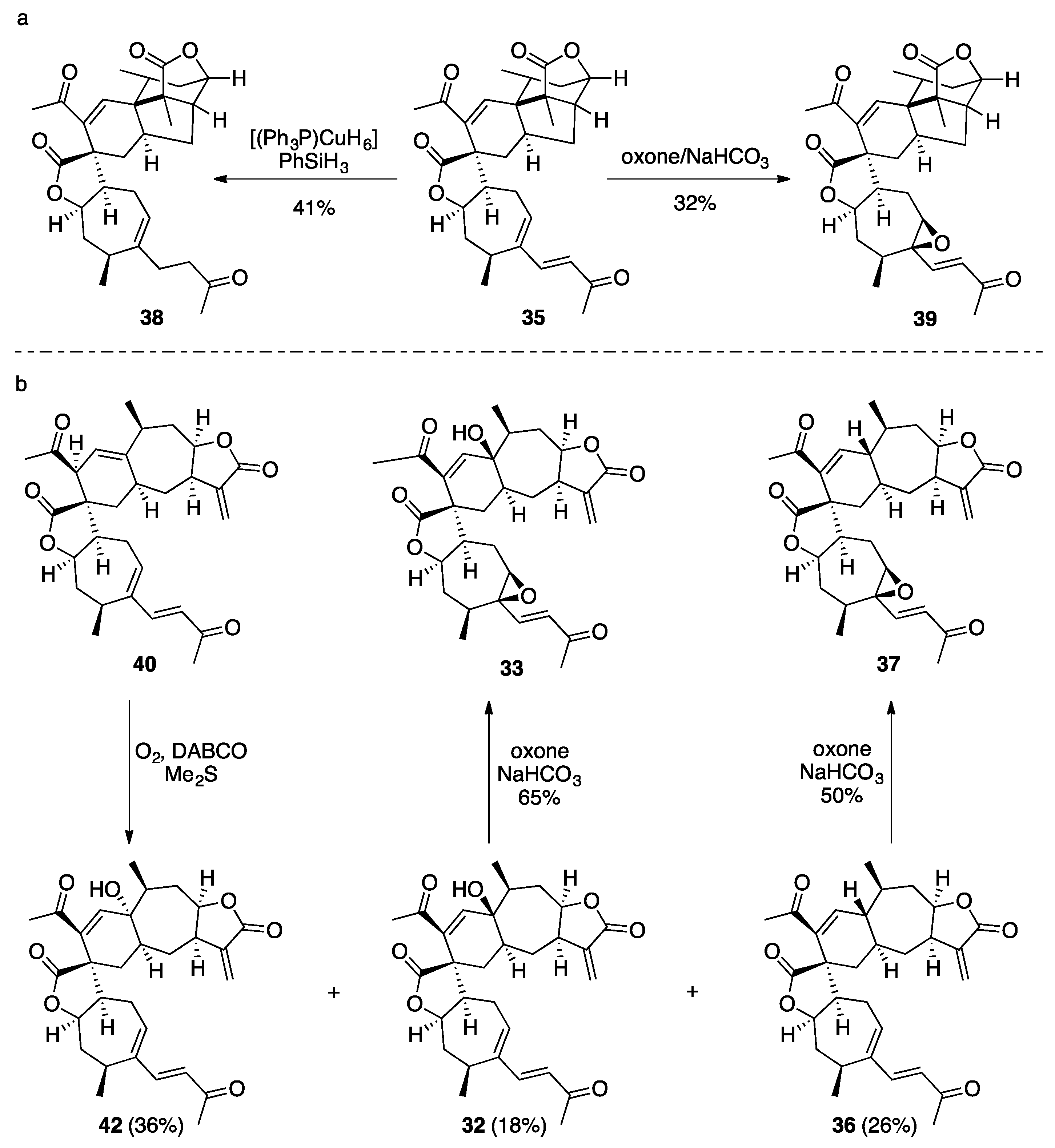
7.1. Pinguisane Type Dimers: Bisacutifolone A and B
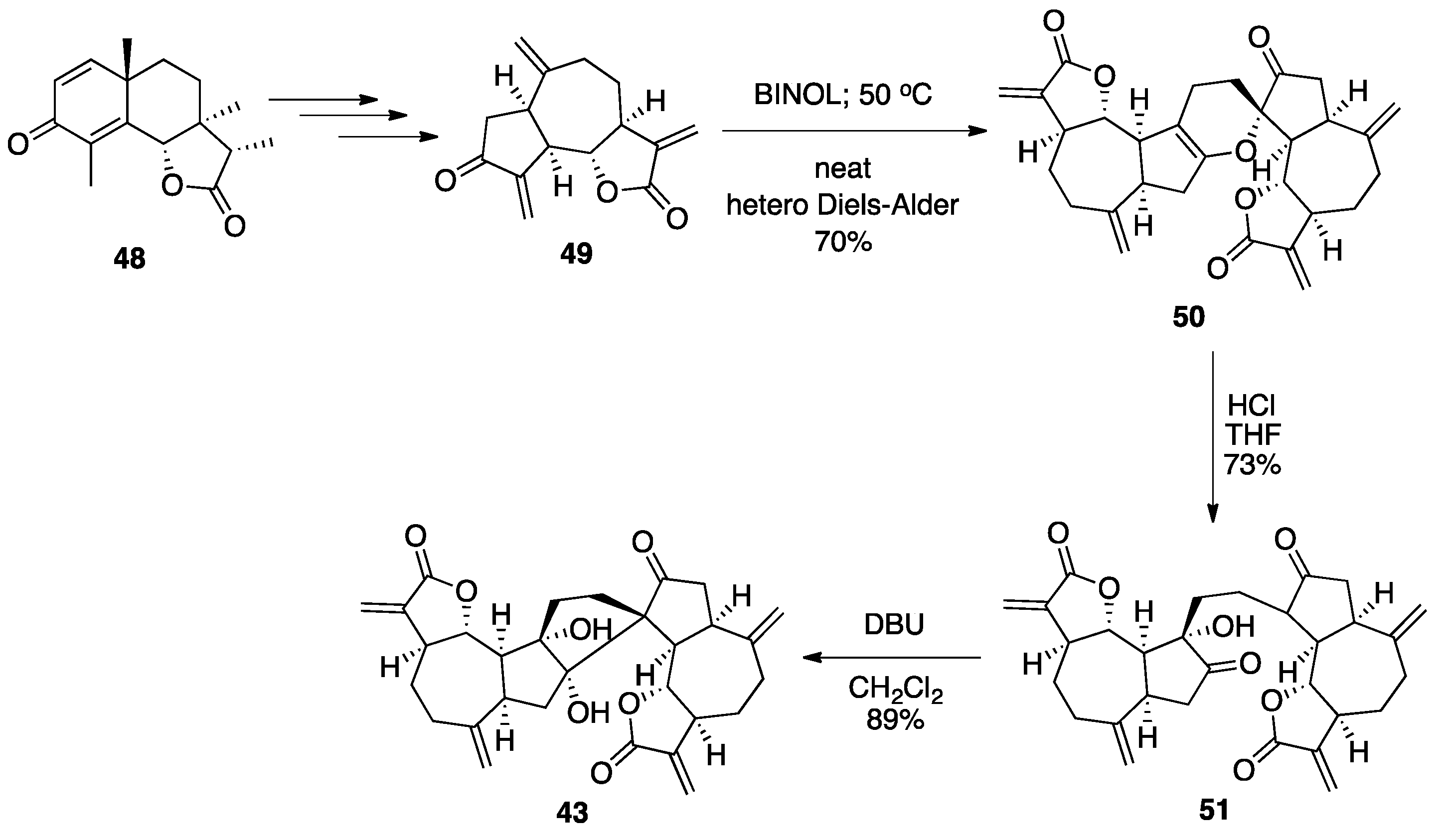
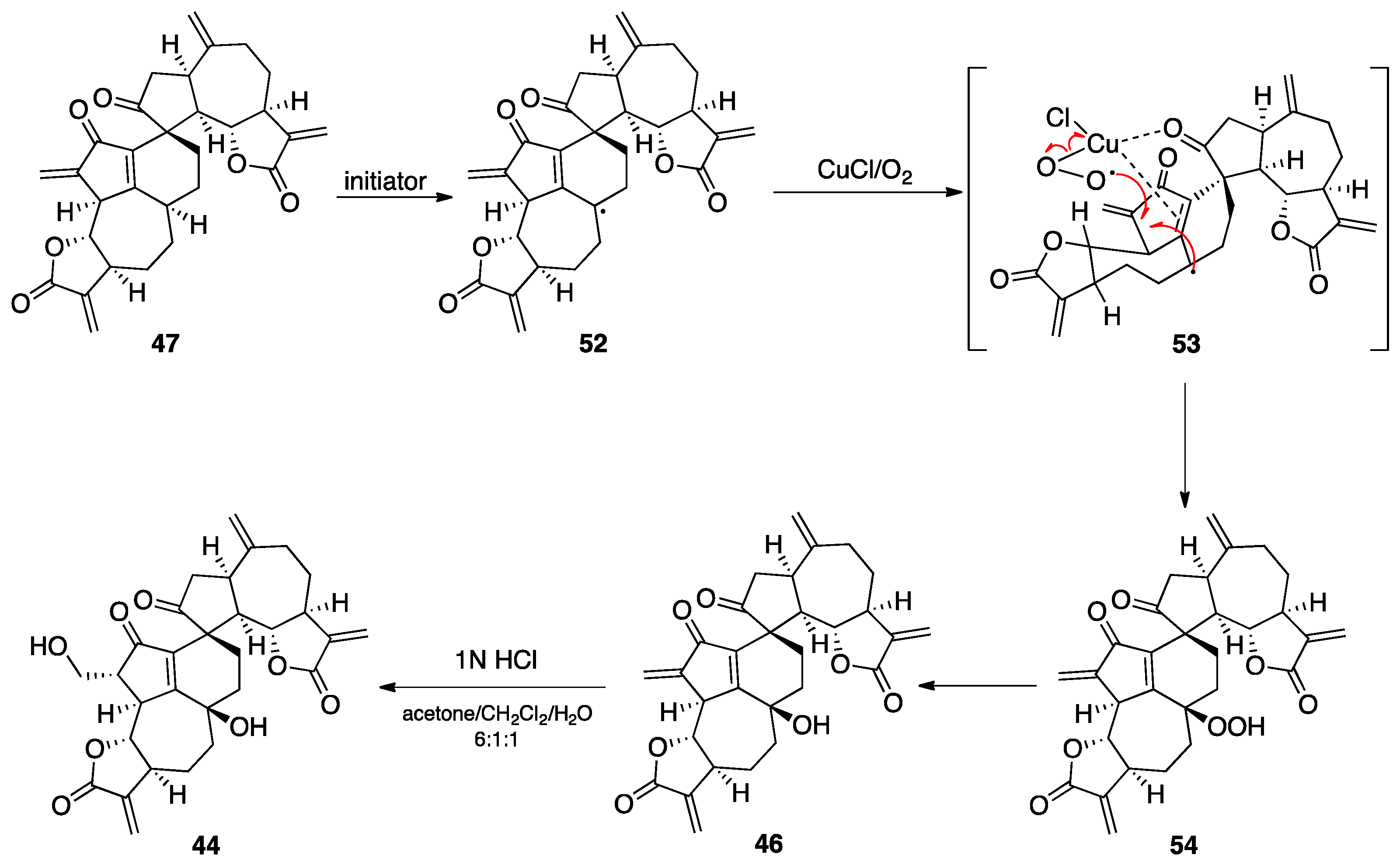
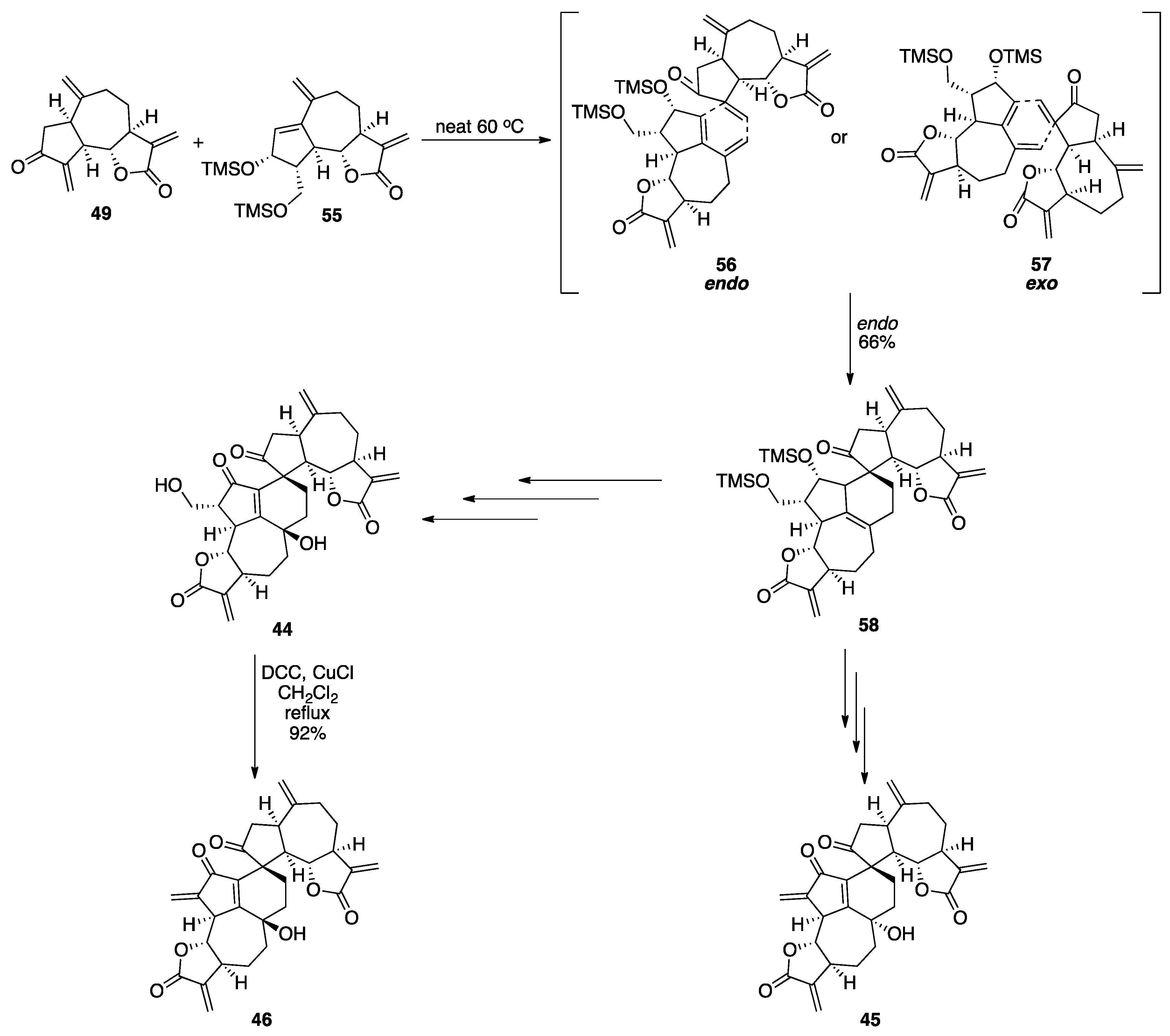
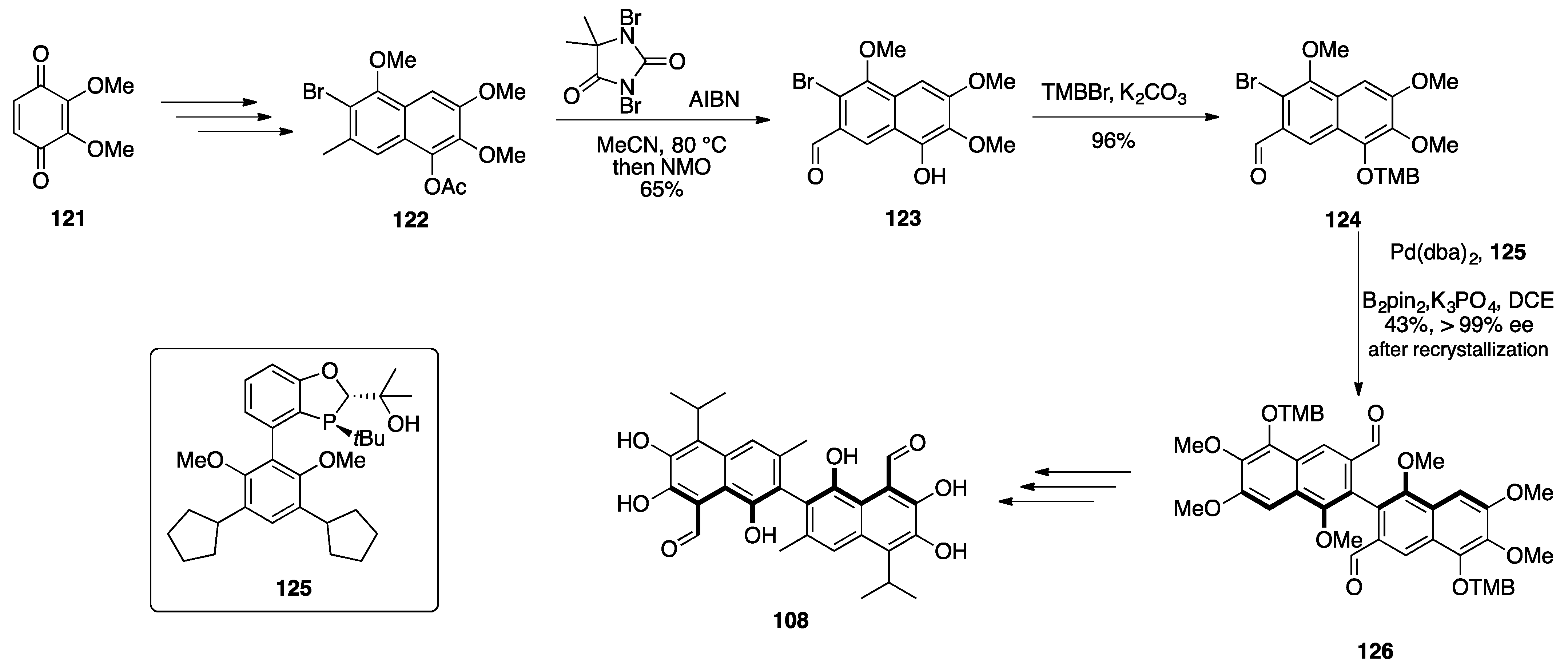
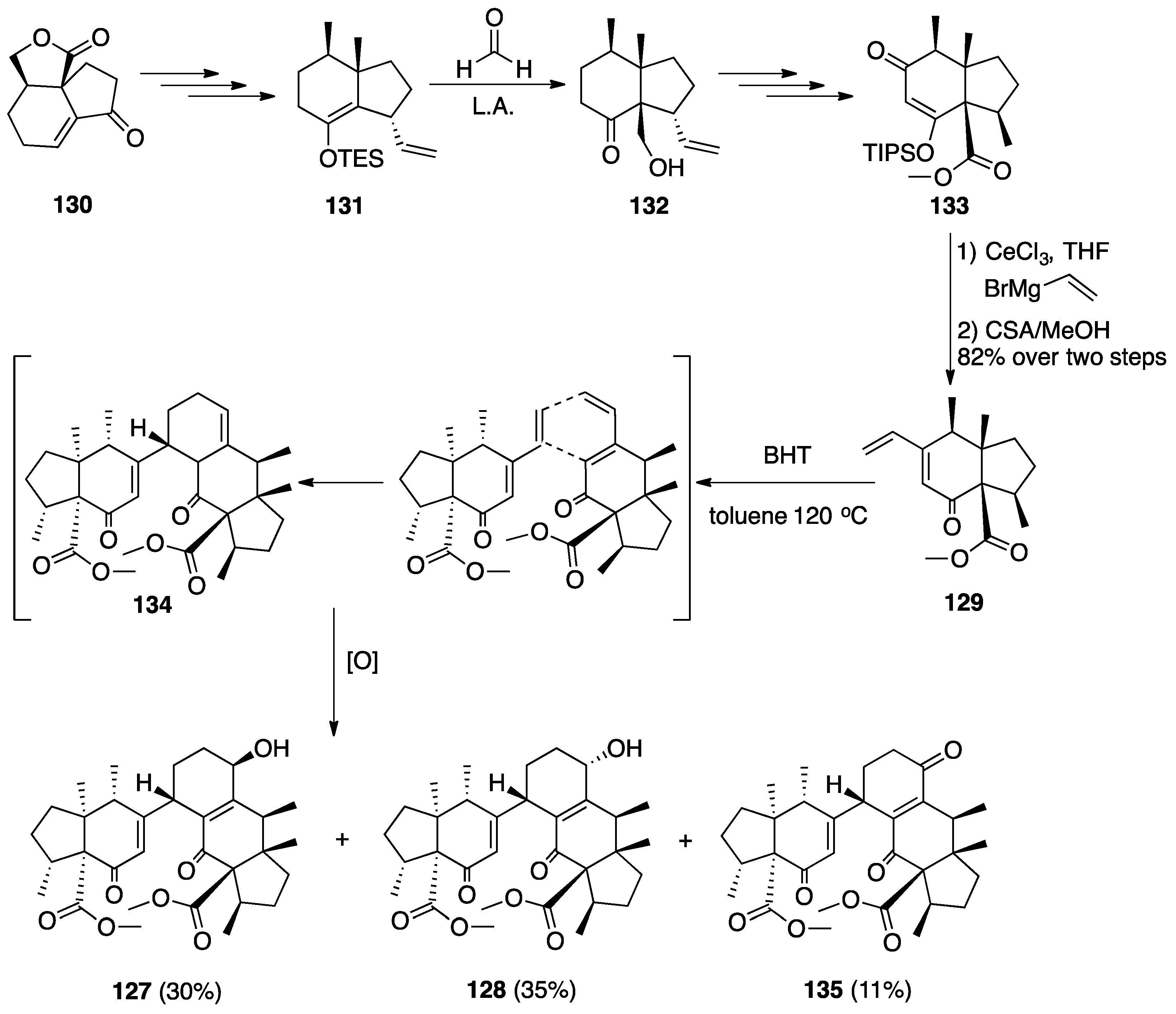
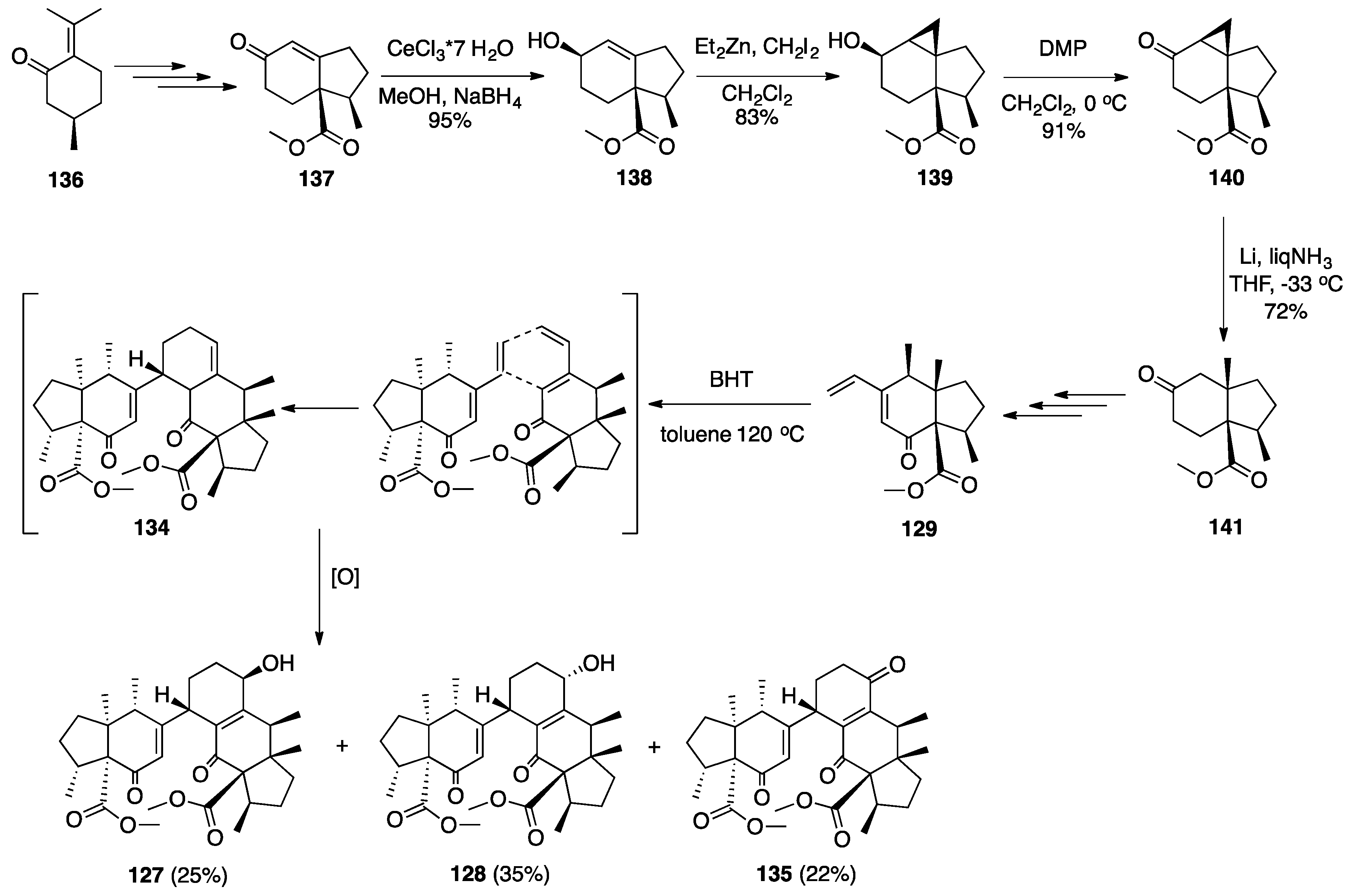
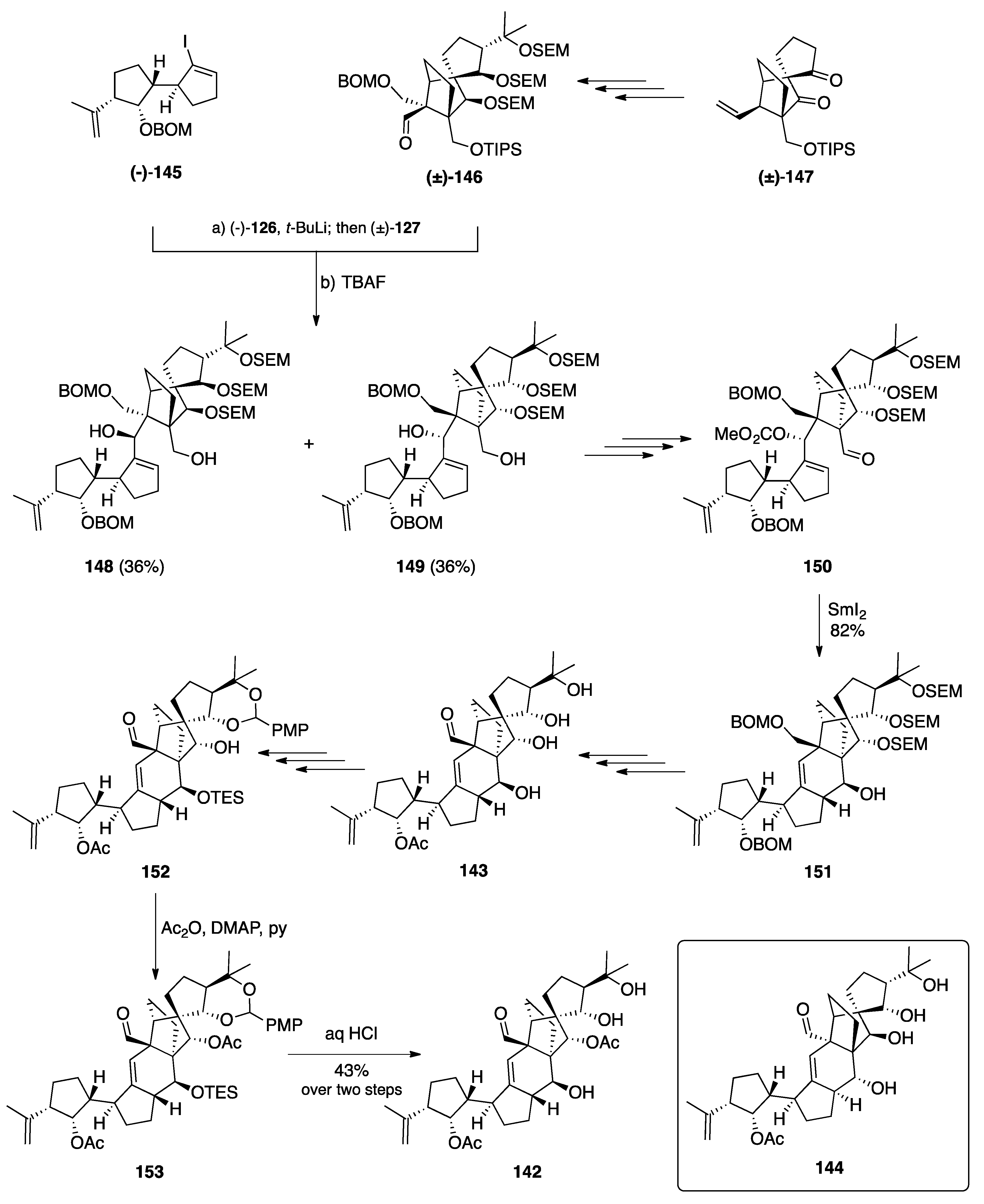
7.2. Vannusals A and B
8. Sesquiterpenoid Alkaloid Dimers
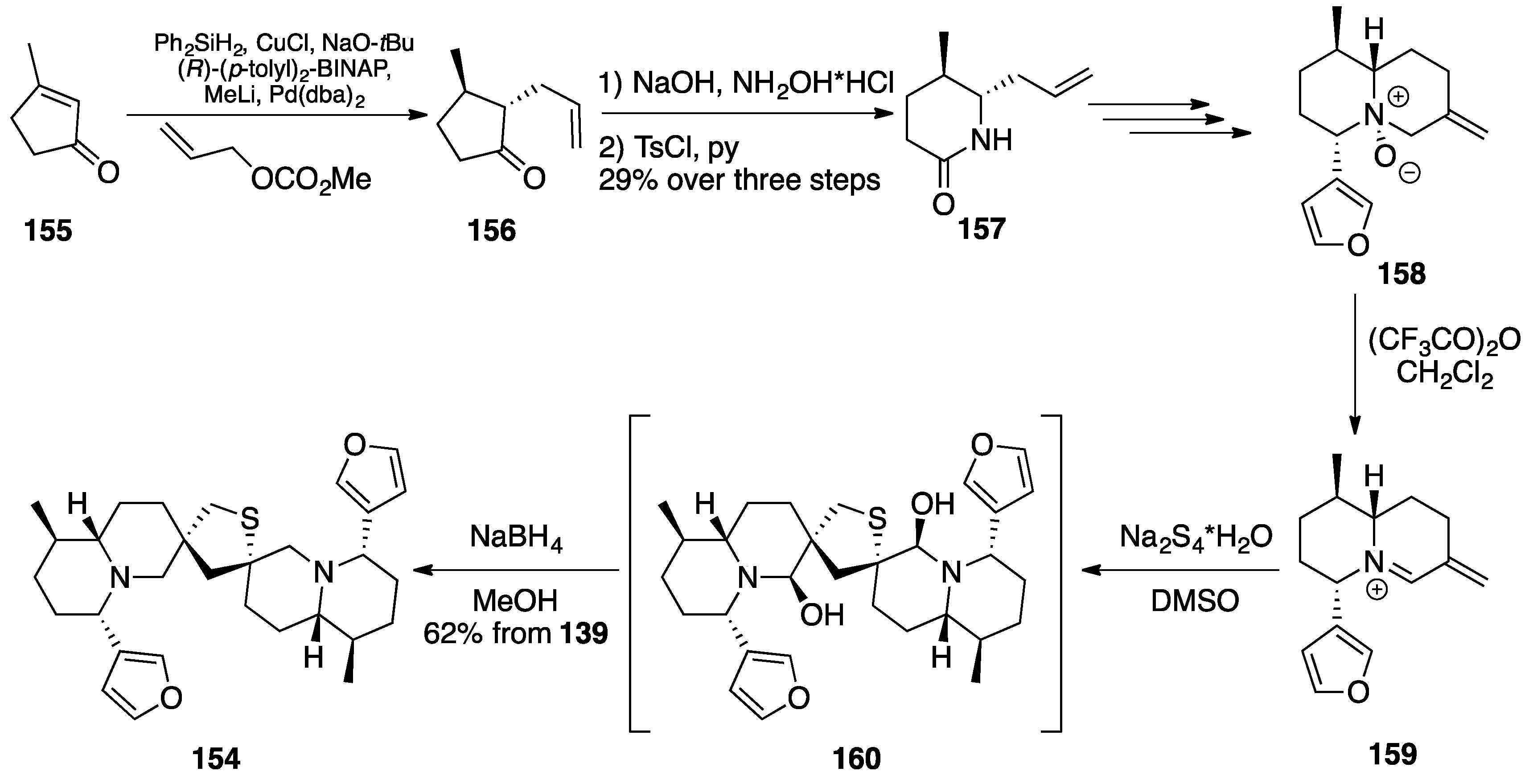
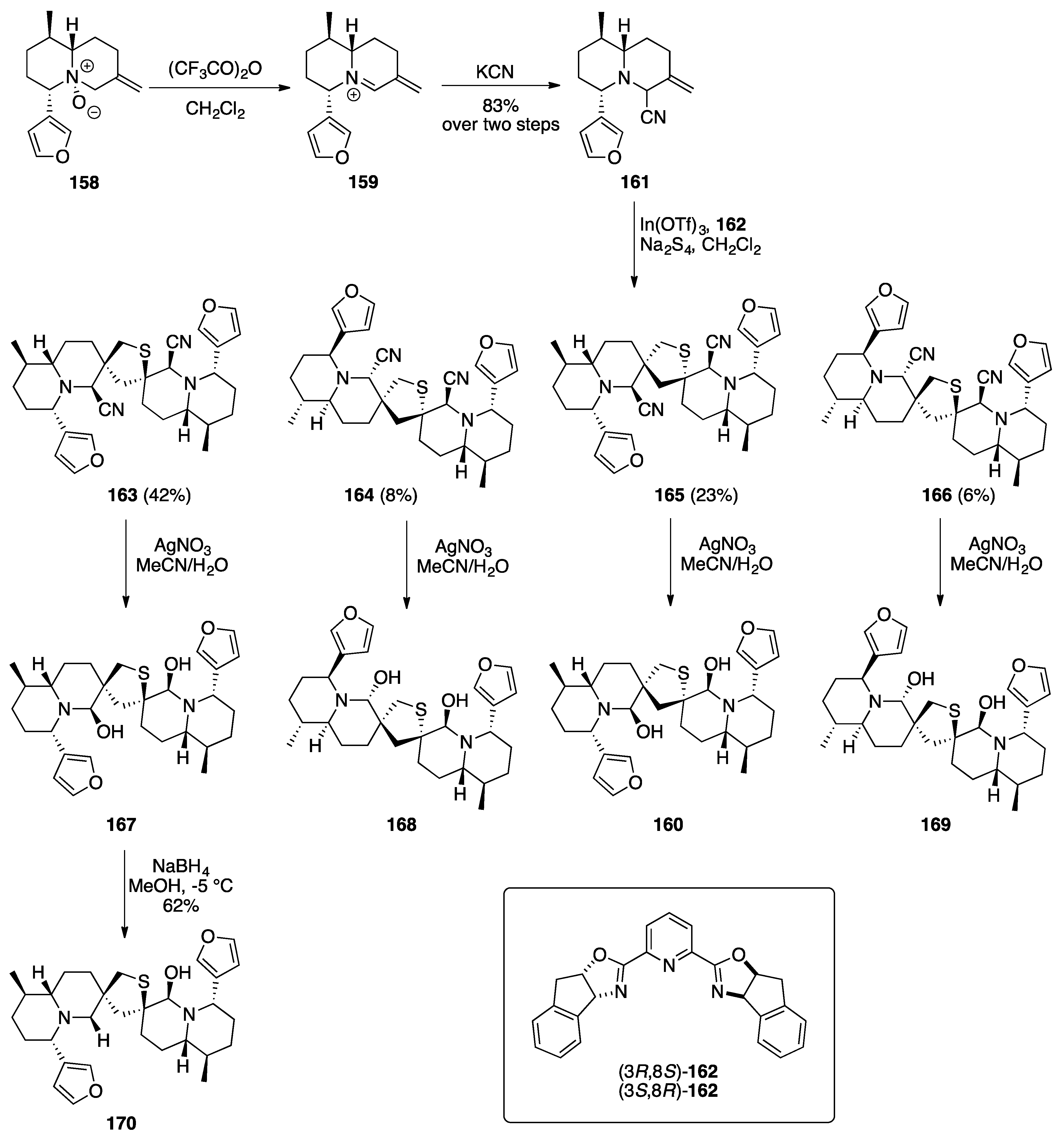
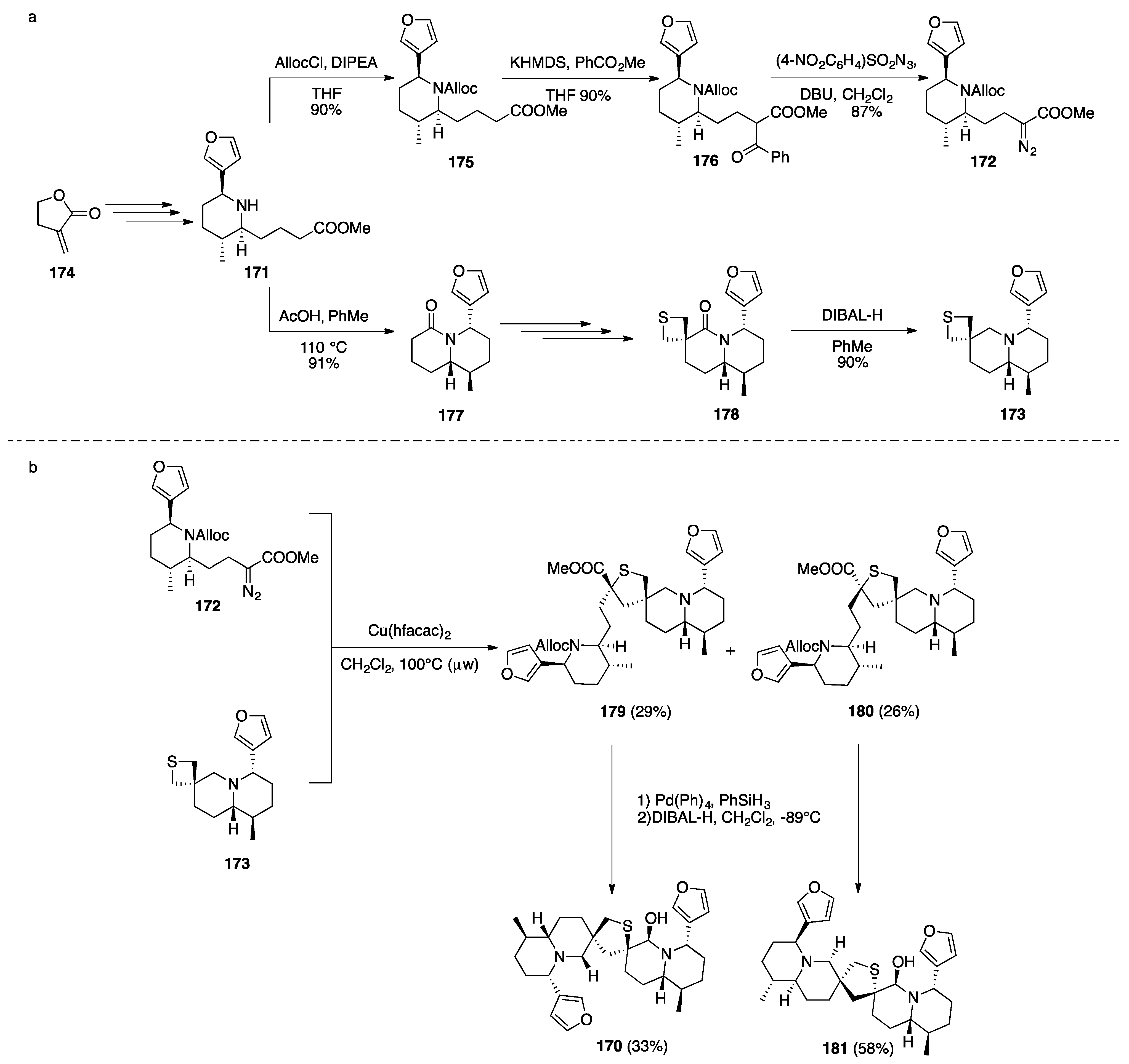
Nuphar Thioalkaloids
9. Merosesquiterpenoid Dimers
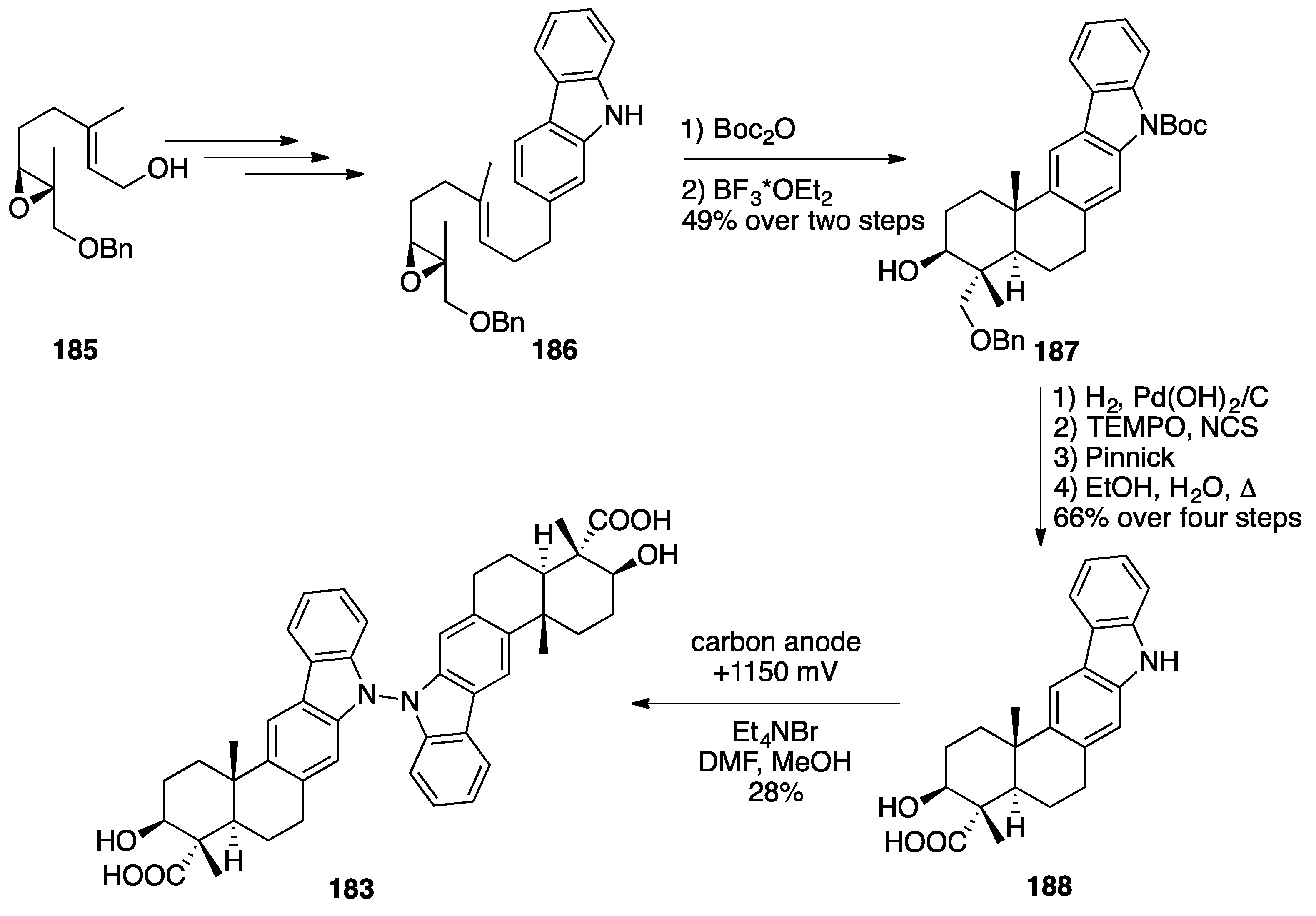
Indolesesquiterpenoid Dimers: Dixiamycin B and Dixiamycin C
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Protecting Groups Abbreviations
References
- Cragg, G.M.; Newman, D.J. Biochimica et Biophysica Acta Natural products: A continuing source of novel drug leads. BBA Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Id, A.M.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Bennett, R.N.; Wallsgrove, R.M. Secondary metabolites in plant defence mechanisms. New Phytol. 1994, 127, 617–633. [Google Scholar] [CrossRef]
- DeGabriel, J.L.; Moore, B.D.; Marsh, K.J.; Foley, W.J. The effect of plant secondary metabolites on the interplay between the internal and external environments of marsupial folivores. Chemoecology 2010, 20, 97–108. [Google Scholar] [CrossRef]
- Yang, L.; Wen, K.S.; Ruan, X.; Zhao, Y.X.; Wei, F.; Wang, Q. Response of plant secondary metabolites to environmental factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef]
- Zhan, Z.J.; Ying, Y.M.; Ma, L.F.; Shan, W.G. Natural disesquiterpenoids. Nat. Prod. Rep. 2011, 28, 594–629. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.H.; Dou, X.X.; Tian, X.H. Natural Disesquiterpenoids: An Overview of Their Chemical Structures, Pharmacological Activities, and Biosynthetic Pathways; Springer: Dordrecht, The Netherlands, 2020; Volume 19, ISBN 0123456789. [Google Scholar]
- Litaudon, M.; Bousserouel, H.; Awang, K.; Nosjean, O.; Martin, M.T.; Dau, M.E.T.H.; Hadi, H.A.; Boutin, J.A.; Sévenet, T.; Guéritte, F. A dimeric sesquiterpenoid from a malaysian meiogyne as a new inhibitor of Bcl-xL/BakBH3 domain peptide interaction. J. Nat. Prod. 2009, 72, 480–483. [Google Scholar] [CrossRef]
- Castilla, C.; Congregado, B.; Chinchón, D.; Torrubia, F.J.; Japón, M.A.; Sáez, C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology 2006, 147, 4960–4967. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Rudin, C.M.; Boise, L.H.; Thompson, C.B. Expression of Bcl-X(L) can confer a multidrug resistance phenotype. Blood 1995, 86, 1903–1910. [Google Scholar] [CrossRef]
- Kharbanda, S.; Pandey, P.; Schofield, L.; Israels, S.; Roncinske, R.; Yoshida, K.; Bharti, A.; Yuan, Z.M.; Saxena, S.; Weichselbaum, R.; et al. Role for BCl-XL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6939–6942. [Google Scholar] [CrossRef]
- Fotsop, D.F.; Roussi, F.; Leverrier, A.; Bretéché, A.; Guéritte, F. Biomimetic total synthesis of meiogynin A, an inhibitor of Bcl-xL and bak interaction. J. Org. Chem. 2010, 75, 7412–7415. [Google Scholar] [CrossRef]
- Stephan, M.; Bernd, L.; Gerald, J.; Roth, H.J.B. An Improved One-pot Procedure for the Synthesis of Alkynes from Aldehydes. Synlett 1996, 6, 521–522. [Google Scholar]
- Lipshutz, B.H.; Butler, T.; Lower, A. Controlling regiochemistry in negishi carboaluminations. Fine tuning the ligand on zirconium. J. Am. Chem. Soc. 2006, 128, 15396–15398. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, L.; Trudell, M.L. Selective oxidation of benzylic alcohols and TBDMS ethers to carbonyl compounds with CrO3-H5IO6. Synthesis 2005, 1757–1760. [Google Scholar] [CrossRef]
- Hagiwara, H.; Okabe, T.; Ono, H.; Kamat, V.P.; Hoshi, T.; Suzuki, T.; Ando, M. Total synthesis of bisabolane sesquiterpenoids, α-bisabol-1-one, curcumene, curcuphenol and elvirol: Utility of catalytic enamine reaction in cyclohexenone synthesis. J. Chem. Soc. Perkin 2002, 2, 895–900. [Google Scholar] [CrossRef]
- Freskos, J.N.; Laneman, S.A.; Reilly, M.L.; Ripin, D.H. Synthesis of chiral succinates via Pd(0) catalyzed carbonylation/asymmetric hydrogenation sequence. Tetrahedron Lett. 1994, 35, 835–838. [Google Scholar] [CrossRef]
- Al-Zoubi, R.M.; Marion, O.; Hall, D.G. Direct and waste-free amidations and cycloadditions by organocatalytic activation of carboxylic acids at room temperature. Angew. Chem. Int. Ed. 2008, 47, 2876–2879. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, J.; Desrat, S.; Guéritte, F.; Roussi, F. Asymmetric Synthesis of Two Analogues of Meiogynin A. Eur. J. Org. Chem. 2013, 2013, 2116–2122. [Google Scholar] [CrossRef]
- Georgantea, P.; Ioannou, E.; Vagias, C.; Roussis, V. Perezoperezone and curcuperezone: Bisabolane dimers from the soft coral Pseudopterogorgia rigida. Tetrahedron Lett. 2013, 54, 6920–6922. [Google Scholar] [CrossRef]
- Long, Y.; Ding, Y.; Wu, H.; Qu, C.; Liang, H.; Zhang, M.; Zhao, X.; Long, X.; Wang, S.; Puno, P.T.; et al. Total Synthesis of (−)-Perezoperezone through an Intermolecular [5+2] Homodimerization of Hydroxy p-Quinone. Angew. Chem. Int. Ed. 2019, 58, 17552–17557. [Google Scholar] [CrossRef]
- Buccini, M.; Punch, K.A.; Kaskow, B.; Flematti, G.R.; Skelton, B.W.; Abraham, L.J.; Piggott, M.J. Ethynylbenzenoid metabolites of Antrodia camphorata: Synthesis and inhibition of TNF expression. Org. Biomol. Chem. 2014, 12, 1100–1113. [Google Scholar] [CrossRef]
- Pinkerton, D.M.; Banwell, M.G.; Willis, A.C. Rapid, chemoenzymatic syntheses of the epoxyquinols ()-bromoxone acetate and ()-tricholomenyn A. Aust. J. Chem. 2009, 62, 1639–1645. [Google Scholar] [CrossRef]
- Ma, X.; Jury, J.C.; Banwell, M.G. Synthesis and reactivity of a putative biogenetic precursor to tricholomenyns B, C, D and E. Tetrahedron Lett. 2011, 52, 2192–2194. [Google Scholar] [CrossRef]
- Li, C.; Chen, T.; Li, B.; Xiao, G.; Tang, W. Efficient synthesis of sterically hindered arenes bearing acyclic secondary alkyl groups by suzuki-miyaura cross-couplings. Angew. Chem. Int. Ed. 2015, 54, 3792–3796. [Google Scholar] [CrossRef]
- Razzak, M.; De Brabander, J.K. Lessons and revelations from biomimetic syntheses. Nat. Chem. Biol. 2011, 7, 865–875. [Google Scholar] [CrossRef]
- Volgraf, M.; Lumb, J.P.; Brastianos, H.C.; Carr, G.; Chung, M.K.W.; Münzel, M.; Mauk, A.G.; Andersen, R.J.; Trauner, D. Biomimetic synthesis of the IDO inhibitors exiguamine A and B. Nat. Chem. Biol. 2008, 4, 535–537. [Google Scholar] [CrossRef]
- Pohnert, G.; Boland, W. Pericyclic reactions in nature: Evidence for a spontaneous [1.7]-hydrogen shift and an 8πe electrocyclic ring closure in the biosynthesis of olefinic hydrocarbons from marine brown algae (phaeophyceae). Tetrahedron 1994, 50, 10235–10244. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Petasis, N.A.; Zipkin, R.E.; Uenishi, J. The Endiandric Acid Cascade. Electrocyclizations in Organic Synthesis. 1. Stepwise, Stereocontrolled Total Synthesis of Endiandric Acids A and B. J. Am. Chem. Soc. 1982, 104, 5555–5557. [Google Scholar] [CrossRef]
- Keller, S.; Nicholson, G.; Drahl, C.; Sorensen, E.; Fiedler, H.P.; Süssmuth, R.D. Abyssomicins G and H and atrop-abyssomicin C from the marine Verrucosispora strain AB-18-032. J. Antibiot. 2007, 60, 391–394. [Google Scholar] [CrossRef]
- Shang, H.; Liu, J.; Bao, R.; Cao, Y.; Zhao, K.; Xiao, C.; Zhou, B.; Hu, L.; Tang, Y. Biomimetic synthesis: Discovery of xanthanolide dimers. Angew. Chem. Int. Ed. 2014, 53, 14494–14498. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ishar, M.P.S. UV irradiation of arylidene-β-ionones in the presence of dioxygen: Regioselective formation of stable endoperoxides. Tetrahedron Lett. 2003, 44, 1943–1945. [Google Scholar] [CrossRef]
- Sharma, V.; Gupta, V.; Anthal, S.; Saxena, A.K.; Ishar, M.P.S. Photochemical formation and decomposition of 8-[β-arylethenyl]-2,2,6- trimethyl-7,9,10-trioxa-tricyclo[6.2.2.01,6]dodec-11-ene to novel 6-hydroxy-1,7,7-trimethyl-2-oxa-bicyclo[4.4.0]dec-4-en-3-one in the presence of oxygen. Tetrahedron Lett. 2012, 53, 5649–5651. [Google Scholar] [CrossRef]
- Dorr, H.; Rawal, V.H. The Intramolecular Diels–Alder Reactions of Photochemically Generated trans-Cycloalkenones. J. Am. Chem. Soc. 1999, 121, 10229–10230. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Loe, Ø.; Stafford, D.G. Sequential cycloaddition approach to the tricyclic core of vibsanin E. Total synthesis of (±)-5-epi-10-epi-vibsanin E. Org. Lett. 2005, 7, 5561–5563. [Google Scholar] [CrossRef]
- Eaton, P.E.; Lin, K. trans-2-Cyclooctenone. J. Am. Chem. Soc. 1964, 86, 2087–2088. [Google Scholar] [CrossRef]
- Corey, E.J.; LaMahieu, M.T.R.; Libit, L. Trans-2-Cycloheptenone. J. Am. Chem. Soc. 1965, 87, 2051–2052. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Venkatesan, K. Photochemical Reactions of Organic Crystals. Chem. Rev. 1987, 87, 433–481. [Google Scholar] [CrossRef]
- Ichikawa, M.; Takahashi, M.; Aoyagi, S.; Kibayashi, C. Total synthesis of (-)-incarvilline, (+)-incarvine C, and (-)-incarvillateine. J. Am. Chem. Soc. 2004, 126, 16553–16558. [Google Scholar] [CrossRef]
- Liu, J.; Wendt, N.L.; Boarman, K.J. Trifluoromethyl groups in crystal design of 1,4-diphenyl-1,3-butadienes for topochemical [2 + 2] photodimerization. Org. Lett. 2005, 7, 1007–1010. [Google Scholar] [CrossRef]
- Otto, S.; Engberts, J.B.F.N. Diels-Alder reactions in water. Pure Appl. Chem. 2000, 72, 1365–1372. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Mahmoud, A.A.; El-Gamal, A.A. A xanthanolide diol and a dimeric xanthanolide from Xanthium species. Planta Med. 1999, 65, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Jakupovic, J.; Bohlmann, F.; Regaila, H.A.; Ahmed, A.M. Sesquiterpene lactones from Xanthium pungens. Phytochemistry 1990, 29, 2211–2215. [Google Scholar] [CrossRef]
- Qu, J.; Deng, S.; Li, L.; Liu, Y.; Li, Y.; Ma, S.; Chen, X.; Yu, S. Cytotoxic dimeric xanthanolides from fruits of Xanthium chinense. Phytochemistry 2016, 132, 115–122. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Li, F.; Liu, X.; Chen, B.; Tang, Y.X.; Wang, M.K. Cytotoxic sesquiterpene lactones from aerial parts of Xanthium sibiricum. Planta Med. 2013, 79, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Nour, A.M.M.; Khalid, S.A.; Kaiser, M.; Brun, R.; Abdallah, W.E.; Schmidt, T.J. The antiprotozoal activity of sixteen asteraceae species native to sudan and bioactivity-guided isolation of xanthanolides from xanthium brasilicum. Planta Med. 2009, 75, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lei, X.; Bao, R.; Li, Y.; Xiao, C.; Hu, L.; Tang, Y. Enantioselective and Collective Total Syntheses of Xanthanolides. Angew. Chem. Int. Ed. 2017, 56, 16323–16327. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Keith, J.; Papa, P.; Vivian, R. A convenient, efficient method for conjugate reductions using catalytic quantities of Cu(I). Tetrahedron Lett. 1998, 39, 4627–4630. [Google Scholar] [CrossRef]
- Ferraz, H.M.C.; Longo, L.S. Bicyclic β-hydroxytetrahydrofurans as precursors of medium ring keto-lactones. J. Org. Chem. 2007, 72, 2945–2950. [Google Scholar] [CrossRef]
- Wu, Z.J.; Xu, X.K.; Shen, Y.H.; Su, J.; Tian, J.M.; Liang, S.; Li, H.A.; Liu, R.H.; Zhang, W.D. Ainsliadimer A, A new sesquiterpene lactone dimer with an unusual carbon skeleton from Ainsliaea macrocephala. Org. Lett. 2008, 10, 2397–2400. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.H.; Jin, H.Z.; Fu, J.J.; Hu, X.J.; Qin, J.J.; Liu, J.H.; Chen, M.; Yan, S.K.; Zhang, W.D. Ainsliatrimers A and B, the first two guaianolide trimers from Ainsliaea fulvioides. Org. Lett. 2008, 10, 5517–5520. [Google Scholar] [CrossRef]
- Bohlmann, F.; Ahmed, M.; Jakupovic, J.; King, R.M.; Robinson, H. Dimeric sesquiterpene lactones and kolavane derivatives from Gochnatia paniculata. Phytochemistry 1983, 22, 191–195. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; A-hirschmann, G.S.; Rlngt, R.M.; Rottinsont, H. Dimeric guaianolides and other constituents from Gochnatia species. Phytochemistry 1986, 25, 1175–1178. [Google Scholar] [CrossRef]
- Li, C.; Yu, X.; Lei, X. A biomimetic total synthesis of (+)-ainsliadimer A. Org. Lett. 2010, 12, 4284–4287. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, W.; Zeng, Z.; Tang, Y. (-)-Gochnatiolide B, synthesized from dehydrocostuslactone, exhibits potent anti-bladder cancer activity in vitro and in vivo. Sci. Rep. 2018, 8, 8807. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, B.R.; Ahuja, N.M.; Bhullar, M.K.; Kalsi, P.S. Some C-3 oxygenated guaianolides from Saussurea lappa. Fitoterapia 1998, 69, 274–275. [Google Scholar]
- Li, C.; Dian, L.; Zhang, W.; Lei, X. Biomimetic syntheses of (-)-gochnatiolides A-C and (-)-ainsliadimer B. J. Am. Chem. Soc. 2012, 134, 12414–12417. [Google Scholar] [CrossRef]
- Deep 2D NMR Studies on Synthetic Gochnatiolide B (46), Led to Structural Reassigment. The Structure was Further Unambiguously Confirmed by X-ray Crystallographic Analysis. Available online: https://pubs.acs.org/doi/suppl/10.1021/ja305464s (accessed on 25 March 2021).
- Xia, D.; Du, Y.; Yi, Z.; Song, H.; Qin, Y. Total syntheses of ainsliadimer B and gochnatiolides A and B. Chem. A Eur. J. 2013, 19, 4423–4427. [Google Scholar] [CrossRef]
- Takeda, Y.; Yamashita, H.; Matsumoto, T.; Terao, H. Chloranthalactone F, A sesquiterpenoid from the leaves of Chloranthus glaber. Phytochemistry 1993, 33, 713–715. [Google Scholar] [CrossRef]
- Okamura, H.; Iwagawa Tetsuo, N.M. A Revised Structure of Chloranthalactone F and Chloranthalactone A Photodimer. Bull. Chem. Soc. Jpn. 1995, 68, 3465–3467. [Google Scholar] [CrossRef]
- Pollini, G.P.; Benetti, S.; De Risi, C.; Zanirato, V. Hagemann’s ester: A timeless building block for natural product synthesis. Tetrahedron 2010, 66, 2775–2802. [Google Scholar] [CrossRef]
- Yue, G.; Yang, L.; Yuan, C.; Du, B.; Liu, B. Total syntheses of lindenane-type sesquiterpenoids: (±)- chloranthalactones A, B, F, (±)-9-hydroxy heterogorgiolide, and (±)-shizukanolide e. Tetrahedron 2012, 68, 9624–9637. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric Synthesis of Bicyclic Intermediates of Natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Qian, S.; Zhao, G. Total synthesis of (+)-chloranthalactone F. Chem. Commun. 2012, 48, 3530–3532. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Ba, M.Y.; Li, X.H.; Guo, J.M.; Qin, X.J.; He, L.; Zhang, Z.Q.; Guo, Y.; Liu, H.Y. Lindenane sesquiterpenoid dimers from Chloranthus japonicus inhibit HIV-1 and HCV replication. Fitoterapia 2016, 115, 64–68. [Google Scholar] [CrossRef]
- Zhou, B.; Wu, Y.; Dalal, S.; Merino, E.F.; Liu, Q.F.; Xu, C.H.; Yuan, T.; Ding, J.; Kingston, D.G.I.; Cassera, M.B.; et al. Nanomolar antimalarial agents against chloroquine-resistant plasmodium falciparum from medicinal plants and their structure-activity relationships. J. Nat. Prod. 2017, 80, 96–107. [Google Scholar] [CrossRef]
- Yang, L.; Yue, G.; Yuan, C.; Du, B.; Deng, H.; Liu, B. Synthetic Studies toward Lindenane-Type Sesquiterpenoid Dimers. Synlett 2014, 25, 2471–2474. [Google Scholar] [CrossRef]
- Wu, J.L.; Lu, Y.S.; Wong, H.N.C.; Peng, X.S. Synthetic studies toward lindenane-type dimers via Diels-Alder reaction. Tetrahedron 2018, 74, 6749–6760. [Google Scholar] [CrossRef]
- Yuan, C.; Du, B.; Deng, H.; Man, Y.; Liu, B. Total Syntheses of Sarcandrolide J and Shizukaol D: Lindenane Sesquiterpenoid [4+2] Dimers. Angew. Chem. Int. Ed. 2017, 56, 637–640. [Google Scholar] [CrossRef]
- Wu, J.L.; Lu, Y.S.; Tang, B.; Peng, X.S. Total syntheses of shizukaols A and E. Nat. Commun. 2018, 9, 4040. [Google Scholar] [CrossRef]
- Du, B.; Huang, Z.; Wang, X.; Chen, T.; Shen, G.; Fu, S.; Liu, B. A unified strategy toward total syntheses of lindenane sesquiterpenoid [4 + 2] dimers. Nat. Commun. 2019, 10, 2–9. [Google Scholar] [CrossRef]
- Longmore, J. Cotton seed oil: Its colouring matter and mucilage, and description of a new method of recovering the loss occurring in the refining process. J. Chem. Ind. 1886, 5, 200–206. [Google Scholar]
- Marchlewski, L. Gossypol, Ein Bestandtheil der Baumwollsamen. J. Prakt Chem. 1899, 60, 84–94. [Google Scholar] [CrossRef]
- Coutinho, E.M. Gossypol: A contraceptive for men. Contraception 2002, 65, 259–263. [Google Scholar] [CrossRef]
- Wang, G.; Nikolovska-coleska, Z.; Yang, C.; Wang, R.; Tang, G.; Guo, J.; Shangary, S.; Qiu, S.; Gao, W.; Yang, D.; et al. Structure-Based Design of Potent Small-Molecule Inhibitors of Anti-Apoptotic Bcl-2 Proteins. J. Med. Chem. Med. 2006, 49, 19–22. [Google Scholar] [CrossRef]
- Meng, Y.; Tang, W.; Dai, Y.; Wu, X.; Liu, M.; Ji, Q.; Ji, M.; Pienta, K.; Lawrence, T.; Xu, L. Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. Cancer Ther. 2008, 7, 2192–2202. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Wang, Q. Regioselective Oxidative Dehydrogenation under Nonenzymatic Conditions: A Synthetic Route to Gossypol. Eur. J. Org. Chem. 2013, 8014–8021. [Google Scholar] [CrossRef]
- Cao, T.; Kong, Y.; Luo, K.; Chen, L.; Zhu, S. Cascade Claisen Rearrangement: Rapid Synthesis of Polysubstituted Salicylaldehydes and Total Syntheses of Hemigossypol and Gossypol. Angew. Chem. Int. Ed. 2018, 57, 8702–8707. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, J.; Gu, W.; Tang, W. Enantioselective Cross-Coupling for Axially Chiral Tetra-ortho-Substituted Biaryls and Asymmetric Synthesis of Gossypol. J. Am. Chem. Soc. 2020, 142, 8036–8043. [Google Scholar] [CrossRef]
- Hashimoto, T.; Irita, H.; Tanaka, M.; Takaoka, S.; Asakawa, Y. Two novel Diels-Alder reaction-type dimeric pinguisane sesquiterpenoids and related compounds from the liverwort Porella acutifolia subsp. tosana. Tetrahedron Lett. 1998, 39, 2977–2980. [Google Scholar] [CrossRef]
- Hashimoto, T.; Irita, H.; Tanaka, M.; Takaoka, S.; Asakawa, Y. Pinguisane and dimeric pinguisane-type sesquiterpenoids from the Japanese liverwort Porella acutifolia subsp. tosana. Phytochemistry 2000, 53, 593–604. [Google Scholar] [CrossRef]
- Shiina, J.; Nishiyama, S. The first total synthesis of acutifolone A, a pinguisane-type sesquiterpenoid isolated from the Japanese liverwort Porella acutifolia subsp. tosana. Tetrahedron Lett. 2005, 46, 7683–7686. [Google Scholar] [CrossRef]
- Hsieh, M.T.; Liu, H.J.; Ly, T.W.; Shia, K.S. A concise total synthesis of (±)-acutifolone A. Org. Biomol. Chem. 2009, 7, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Shiina, J.; Oikawa, M.; Nakamura, K.; Obata, R.; Nishiyama, S. Synthesis of pinguisane-type sesquiterpenoids acutifolone A, pinguisenol, and bisacutifolones by a Diels-Alder dimerization reaction. Eur. J. Org. Chem. 2007, 5190–5197. [Google Scholar] [CrossRef]
- Johnsson, L.; Andersson, R.E.; Dutta, P.C. Side-chain autoxidation of stigmasterol and analysis of a mixture of phytosterol oxidation products by chromatographic and spectroscopic methods. JAOCS J. Am. Oil Chem. Soc. 2003, 80, 777–783. [Google Scholar] [CrossRef]
- Johnsson, L.; Dutta, P.C. Characterization of side-chain oxidation products of sitosterol and campesterol by chromatographic and spectroscopic methods. JAOCS J. Am. Oil Chem. Soc. 2003, 80, 767–776. [Google Scholar] [CrossRef]
- Yadav, J.S.; Singh, S.; Das, S. Stereoselective Total Syntheses of Acutifolone A, Bisacutifolone A and B, Pinguisenol, and Isonaviculol. ACS Omega 2018, 3, 636–647. [Google Scholar] [CrossRef]
- Guella, G.; Dini, F.; Pietra, F. Metabolites with a novel C30 backbone from marine ciliates. Angew. Chem. Int. Ed. 1999, 38, 1134–1136. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Zhang, H.; Ortiz, A.; Dagneau, P. Total synthesis of the originally assigned structure of vannusal B. Angew. Chem. Int. Ed. 2008, 47, 8605–8610. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Dagneau, P.; Lanver, A.; Jennings, M.P.; Arseniyadis, S.; Faraoni, R.; Lizos, D.E. Total synthesis and structural revision of vannusals A and B: Synthesis of the originally assigned structure of vannusal B. J. Am. Chem. Soc. 2010, 132, 7138–7152. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Guella, G. Total synthesis and structural revision of vannusals A and B: Synthesis of the true structures of vannusals A and B. J. Am. Chem. Soc. 2010, 132, 7153–7176. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Tang, W.; Dagneau, P.; Faraoni, R. A catalytic asymmetric three-component 1,4-addition/aldol reaction: Enantioselective synthesis of the spirocyclic system of vannusal A. Angew. Chem. Int. Ed. 2005, 44, 3874–3879. [Google Scholar] [CrossRef]
- Lalonde, R.T.; Wong, C. properties of sulfur containing nuphar alkaloids. Pure Appl. Chem. 1977, 49, 169–181. [Google Scholar] [CrossRef]
- Matsuda, H.; Morikawa, T.; Oda, M.; Asao, Y.; Yoshikawa, M. Potent anti-metastatic activity of dimeric sesquiterpene thioalkaloids from the rhizome of Nuphar pumilum. Bioorg. Med. Chem. Lett. 2003, 13, 4445–4449. [Google Scholar] [CrossRef] [PubMed]
- Ozer, J.; Eisner, N.; Ostrozhenkova, E.; Bacher, A.; Eisenreich, W.; Benharroch, D.; Golan-Goldhirsh, A.; Gopas, J. Nuphar lutea thioalkaloids inhibit the nuclear factor κB pathway, potentiate apoptosis and are synergistic with cisplatin and etoposide. Cancer Biol. Ther. 2009, 8, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Jansen, D.J.; Shenvi, R.A. Synthesis of (−)-Neothiobinupharidine. J. Am. Chem. Soc. 2013, 135, 10–13. [Google Scholar] [CrossRef]
- Korotkov, A.; Li, H.; Chapman, C.W.; Xue, H.; Macmillan, J.B.; Eastman, A.; Wu, J. Total Syntheses and Biological Evaluation of Both Enantiomers of Several Hydroxylated Dimeric Nuphar Alkaloids. Angew. Chem. Int. Ed. 2015, 54, 10604–10607. [Google Scholar] [CrossRef]
- Lacharity, J.J.; Fournier, J.; Lu, P.; Mailyan, A.K.; Herrmann, A.T.; Zakarian, A. Total Synthesis of Unsymmetrically Oxidized Nuphar Thioalkaloids via Copper-Catalyzed Thiolane Assembly. J. Am. Chem. Soc. 2017, 139, 13272–13275. [Google Scholar] [CrossRef]
- Marcos, I.S.; Moro, R.F.; Costales, I.; Basabe, P.; Díez, D. Sesquiterpenyl indoles. Nat. Prod. Rep. 2013, 30, 1509–1526. [Google Scholar] [CrossRef]
- Ding, L.; Münch, J.; Goerls, H.; Maier, A.; Fiebig, H.H.; Lin, W.H.; Hertweck, C. Xiamycin, a pentacyclic indolosesquiterpene with selective anti-HIV activity from a bacterial mangrove endophyte. Bioorg. Med. Chem. Lett. 2010, 20, 6685–6687. [Google Scholar] [CrossRef]
- Ding, L.; Maier, A.; Fiebig, H.H.; Lin, W.H.; Hertweck, C. A family of multicyclic indolosesquiterpenes from a bacterial endophyte. Org. Biomol. Chem. 2011, 9, 4029–4031. [Google Scholar] [CrossRef]
- Zhang, Q.; Mándi, A.; Li, S.; Chen, Y.; Zhang, W.; Tian, X.; Zhang, H.; Li, H.; Zhang, W.; Zhang, S.; et al. N-N-coupled indolo-sesquiterpene atropo-diastereomers from a marine-derived actinomycete. Eur. J. Org. Chem. 2012, 5256–5262. [Google Scholar] [CrossRef]
- Xu, Z.; Baunach, M.; Ding, L.; Hertweck, C. Bacterial synthesis of diverse indole terpene alkaloids by an unparalleled cyclization sequence. Angew. Chem. Int. Ed. 2012, 51, 10293–10297. [Google Scholar] [CrossRef] [PubMed]
- Baunach, M.; Ding, L.; Bruhn, T.; Bringmann, G.; Hertweck, C. Regiodivergent N-C and N-N aryl coupling reactions of indoloterpenes and cycloether formation mediated by a single bacterial flavoenzyme. Angew. Chem. Int. Ed. 2013, 52, 9040–9043. [Google Scholar] [CrossRef]
- Rosen, B.R.; Werner, E.W.; O’Brien, A.G.; Baran, P.S. Total synthesis of dixiamycin B by electrochemical oxidation. J. Am. Chem. Soc. 2014, 136, 5571–5574. [Google Scholar] [CrossRef]
- Forero-Cortés, P.A.; Haydl, A.M. The 25th Anniversary of the Buchwald-Hartwig Amination: Development, Applications, and Outlook. Org. Process. Res. Dev. 2019, 23, 1478–1483. [Google Scholar] [CrossRef]
- Meng, Z.; Yu, H.; Li, L.; Tao, W.; Chen, H.; Wan, M.; Yang, P.; Edmonds, D.J.; Zhong, J.; Li, A. Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nat. Commun. 2015, 6, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Antilla, J.C.; Huang, X.; Buchwald, S.L.; May, R. V A General and Efficient Copper Catalyst for the Amidation of Aryl Halides and the N -Arylation of Nitrogen Heterocycles. J. Am. Chem. Soc. 2001, 7727–7729. [Google Scholar] [CrossRef] [PubMed]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Conditions | 45 a | 46 a | 47 a |
| Pd(OAc)2/DMSO/air/50 °C | 40 | 6 | 9 |
| Pd(OAc)2/DMSO/glovebox/50 °C | 0 | 0 | 14 |
| Pd(OAc)2/DMSO/CuCl/air/50 °C | 6 | 27 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caprioglio, D.; Salamone, S.; Pollastro, F.; Minassi, A. Biomimetic Approaches to the Synthesis of Natural Disesquiterpenoids: An Update. Plants 2021, 10, 677. https://doi.org/10.3390/plants10040677
Caprioglio D, Salamone S, Pollastro F, Minassi A. Biomimetic Approaches to the Synthesis of Natural Disesquiterpenoids: An Update. Plants. 2021; 10(4):677. https://doi.org/10.3390/plants10040677
Chicago/Turabian StyleCaprioglio, Diego, Stefano Salamone, Federica Pollastro, and Alberto Minassi. 2021. "Biomimetic Approaches to the Synthesis of Natural Disesquiterpenoids: An Update" Plants 10, no. 4: 677. https://doi.org/10.3390/plants10040677
APA StyleCaprioglio, D., Salamone, S., Pollastro, F., & Minassi, A. (2021). Biomimetic Approaches to the Synthesis of Natural Disesquiterpenoids: An Update. Plants, 10(4), 677. https://doi.org/10.3390/plants10040677