
Evolutionary Divergence and Biased Expression of NAC Transcription Factors in Hexaploid Bread Wheat (Triticum aestivum L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification of NAC Transcription Factors in Green Plants
2.2. Genome-Wide Identification of the NAC Transcription Factors from T. aestivum
2.3. The Evolutionary Process of the NAC Transcription Factors from Diploid Species to Hexaploid Wheat
2.4. Genome Synteny Analysis of TaNACs
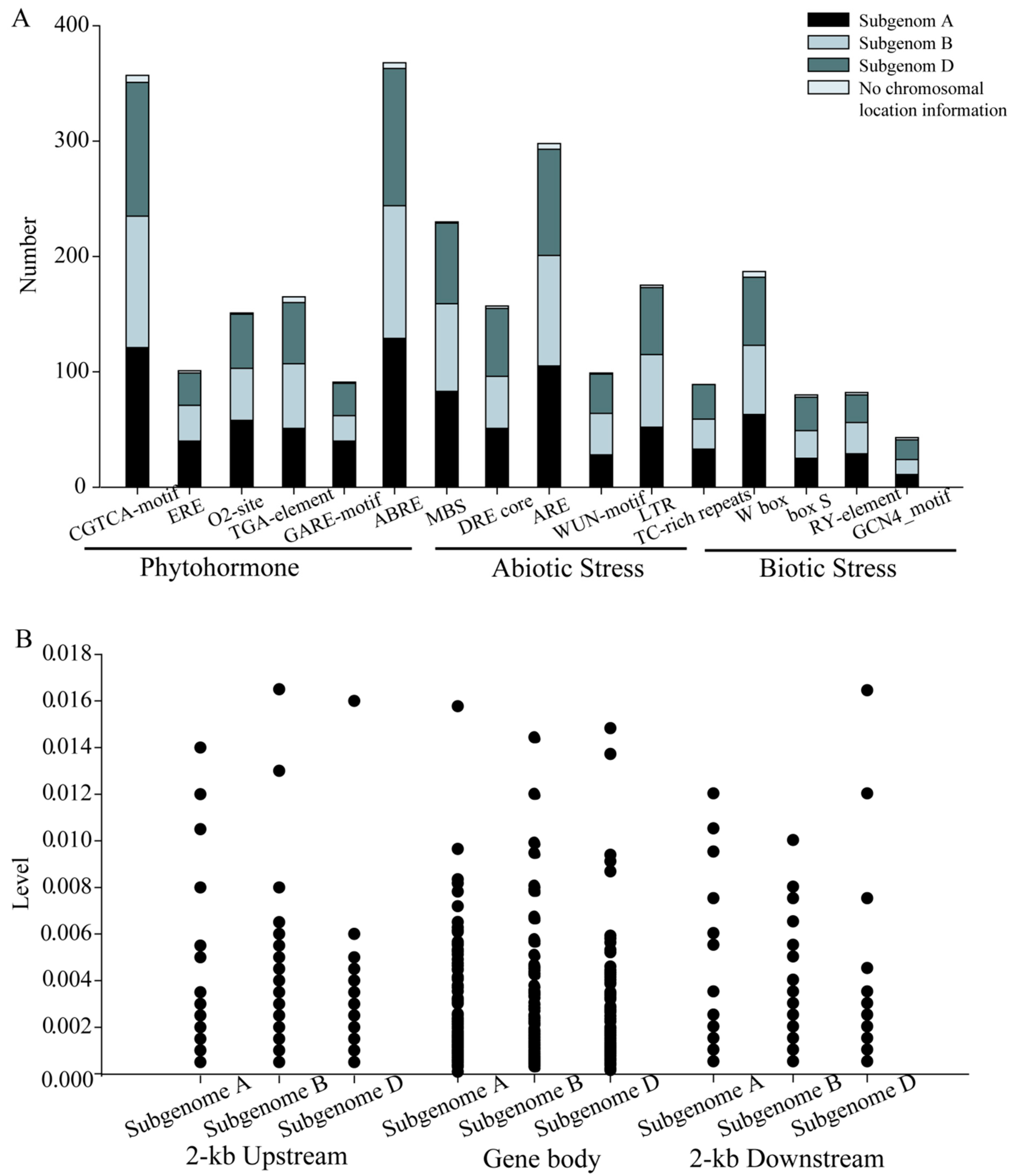
2.5. Cis-Component and SNP Analysis of the NAC Family Genes in T. aestivum
2.6. Biased Expression of TaNACs in Response to Abiotic Stress
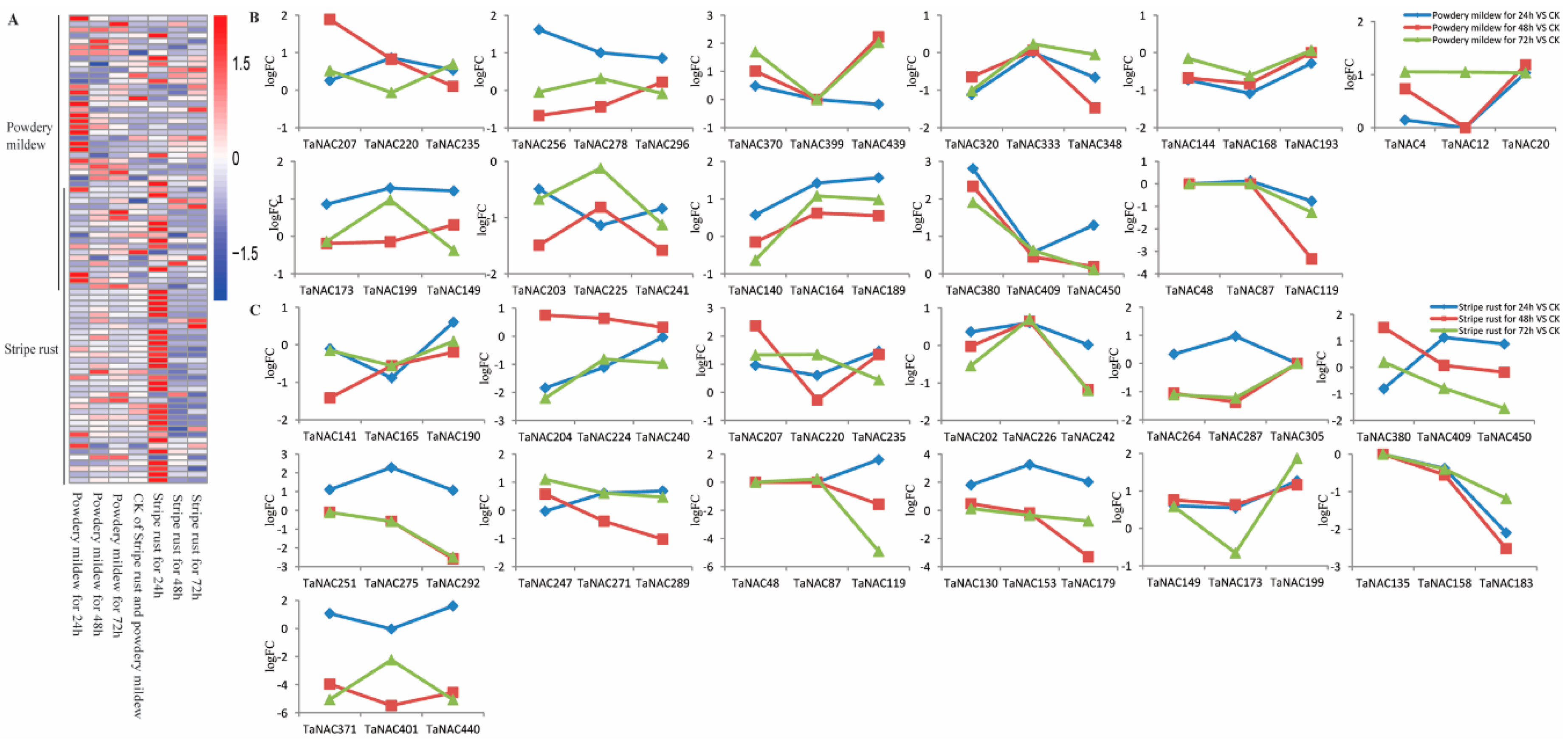
2.7. Biased Expression of TaNACs in Response to Biotic Stress
3. Discussion
4. Materials and Methods
4.1. Identification of NAC Transcription Factor from T. aestivum and Other Green Plants
4.2. Cloning and Transactivation Activity Assay of TaNAC140, and TaNAC290
4.3. Gene Structure and Phylogenetic Analysis
4.4. Chromosomal Location and Collinearity Analysis
4.5. Cis-Element Analysis
4.6. Transcriptome Data and Gene Expression Pattern Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Souer, E.; Houwelingen, A.; Kloos, D.; Mol, J.; Koes, R. The no apical meristem gene of petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell 1996, 85, 159–170. [Google Scholar] [CrossRef]
- Aida, M.; Ishida, T.; Fukaki, H.; Fujisawa, H.; Tasaka, M. Genes involved in organ separation in Arabidopsis: An analysis of the cup-shaped cotyledon mutant. Plant Cell 1997, 9, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, D.; Zhang, H.; Song, F. Functions of NAC transcription factors in biotic and abiotic stress responses in plants. Yi Chuan 2012, 34, 993–1002. [Google Scholar] [CrossRef]
- Ooka, H.; Satoh, K.; Doi, K.; Nagata, T.; Otomo, Y.; Murakami, K.; Matsubara, K.; Osato, N.; Kawai, J.; Carninci, P.; et al. Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 2003, 10, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Ernst, H.A.; Olsen, A.N.; Skriver, K.; Larsen, S.; Leggio, L.L. Structure of the conserved domain of ANAC, a member of the NAC family of transcription factors. EMBO Rep. 2004, 5, 297–303. [Google Scholar] [CrossRef]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Puranik, S.; Sahu, P.P.; Srivastava, P.S.; Prasad, M. NAC proteins: Regulation and role in stress tolerance. Trends Plant Sci. 2012, 17, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, S.G.; Kim, Y.S.; Seo, P.J.; Bae, M.; Yoon, H.K.; Park, C.M. Exploring membrane-associated NAC transcription factors in Arabidopsis: Implications for membrane biology in genome regulation. Nucleic Acids Res. 2007, 35, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef]
- Jensen, M.K.; Skriver, K. NAC transcription factor gene regulatory and protein-protein interaction networks in plant stress responses and senescence. IUBMB Life 2014, 66, 156–166. [Google Scholar] [CrossRef]
- Erpen, L.; Devi, H.S.; Grosser, J.W.; Dutt, M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tissue Organ Cult. 2017, 132, 1–25. [Google Scholar] [CrossRef]
- Sun, H.; Hu, M.; Li, J.; Chen, L.; Li, M.; Zhang, S.; Zhang, X.; Yang, X. Comprehensive analysis of NAC transcription factors uncovers their roles during fiber development and stress response in cotton. BMC Plant Biol. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Manimekalai, R.; Sharoni, A.M.; Satoh, K.; Kondoh, H.; Ooka, H.; Kikuchi, S. Genome-wide analysis of NAC transcription factor family in rice. Gene 2010, 465, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, K.; El-Kereamy, A.; Kim, S.H.; Nambara, E.; Rothstein, S.J. ANAC032 positively regulates age-dependent and stress-induced senescence in Arabidopsis thaliana. Plant Cell Physiol. 2016, 57, 2029–2046. [Google Scholar] [CrossRef] [PubMed]
- Gladman, N.P.; Marshall, R.S.; Lee, K.H.; Vierstra, R.D. The proteasome stress regulon is controlled by a pair of NAC transcription factors in Arabidopsis. Plant Cell 2016, 28, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Shahnejat-Bushehri, S.; Tarkowska, D.; Sakuraba, Y.; Balazadeh, S. Arabidopsis NAC transcription factor JUB1 regulates GA/BR metabolism and signalling. Nat. Plants 2016, 29, 16013. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Piao, W.; Lim, J.-H.; Han, S.-H.; Kim, Y.-S.; An, G.; Paek, N.-C. Rice onac106 inhibits leaf senescence and increases salt tolerance and tiller angle. Plant Cell Physiol. 2015, 56, 2325–2339. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Oh, N.; Chung, P.J.; Kim, Y.S.; Choi, Y.D.; Kim, J.-K. Overexpression of OsNAC14 Improves Drought Tolerance in Rice. Front. Plant Sci. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Huang, L.; Hong, Y.; Zhang, H.; Song, F.; Li, D. Comprehensive analysis suggests overlapping expression of rice ONAC transcription factors in abiotic and biotic stress responses. Int. J. Mol. Sci. 2015, 16, 4306–4326. [Google Scholar] [CrossRef]
- Christiansen, M.W.; Holm, P.B.; Gregersen, P.L. Characterization of barley (Hordeum vulgare L.) NAC transcription factors suggests conserved functions compared to both monocots and dicots. BMC Res. Notes 2011, 19, 302. [Google Scholar] [CrossRef]
- Murozuk, E.; Massange-Sánchez, J.A.; Nielsen, K.; Gregersen, P.L.; Braumann, I. Genome wide characterization of barley NAC transcription factors enables the identification of grain-specific transcription factors exclusive for the Poaceae family of monocotyledonous plants. PLoS ONE 2018, 13, e0209769. [Google Scholar] [CrossRef] [PubMed]
- Saidi, M.N.; Mergby, D.; Brini, F. Identification and expression analysis of the NAC transcription factor family in durum wheat (Triticum turgidum L. ssp. durum). Plant Physiol. Biochem. 2017, 112, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Borrill, P.; Harrington, S.A.; Uauy, C. Genome-wide sequence and expression analysis of the NAC transcription factor family in polyploid wheat. Genes 2017, 7, 3019–3029. [Google Scholar] [CrossRef]
- Distelfeld, A.; Pearce, S.P.; Avni, R.; Scherer, B.; Uauy, C.; Piston, F.; Slade, A.; Zhao, R.; Dubcovsky, J. Divergent functions of orthologous NAC transcription factors in wheat and rice. Plant Mol. Biol. 2012, 78, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Khan, A.A.; Lin, Q.; Han, Y.; Mu, P.; Liu, Y.; Zhang, H.; Li, L.; Meng, X.; et al. Expression partitioning of homeologs and tandem duplications contribute to salt tolerance in wheat (Triticum aestivum L.). Sci. Rep. 2016, 6, 21476. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef]
- Huu, C.N.; Keller, B.; Conti, E.; Kappel, C. Supergene evolution via stepwise duplications and neofunctionalization of a floral-organ identity gene. Proc. Natl. Acad. Sci. USA 2020, 117, 23148–23157. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 7191. [Google Scholar]
- Zhao, D.; Derkx, A.P.; Liu, D.C.; Buchner, P.; Hawkesford, M.J.; Flemetakis, E. Overexpression of a NAC transcription factor delays leaf senescence and increases grain nitrogen concentration in wheat. Plant Biol. 2015, 17, 904–913. [Google Scholar] [CrossRef]
- Feng, H.; Duan, X.; Zhang, Q.; Li, X.; Wang, B.; Huang, L.; Wang, X.; Kang, Z. The target gene of tae-miR164, a novel NAC transcription factor from the NAM subfamily, negatively regulates resistance of wheat to stripe rust. Mol. Plant Pathol. 2014, 15, 284–296. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zou, C.; Li, K.; Wang, K.; Li, T.; Gao, L.; Zhang, X.; Wang, H.; Yang, Z.; Liu, X.; et al. The Aegilops tauschii genome reveals multiple impacts of transposons. Nat. Plants 2017, 3, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.-Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef]
- Singh, N.K.; Anand, S.; Jain, A.; Das, S. Comparative genomics and synteny analysis of KCS17-KCS18 cluster across different genomes and sub-genomes of brassicaceae for analysis of its evolutionary history. Plant Mol. Biol. Rep. 2016, 35, 237–251. [Google Scholar] [CrossRef]
- He, F.; Pasam, R.; Shi, F.; Kant, S.; Keeble-Gagnere, G.; Kay, P.; Forrest, K.; Fritz, A.; Hucl, P.; Wiebe, K.; et al. Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat. Genet. 2019, 51, 896–904. [Google Scholar] [CrossRef]
- Hao, C.; Jiao, C.; Hou, J.; Li, T.; Liu, H.; Wang, Y.; Zheng, J.; Liu, H.; Bi, Z.; Xu, F.; et al. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol. Plant 2020, 13, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Wang, H.; Tang, X. NAC transcription factors in plant multiple abiotic stress responses: Progress and prospects. Front. Plant Sci. 2015, 6, 902. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xin, M.; Qin, J.; Peng, H.; Ni, Z.; Yao, Y.; Sun, Q. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 2015, 15, 152. [Google Scholar] [CrossRef]
- Li, C.; Zhang, W.; Yuan, M.; Jiang, L.; Sun, B.; Zhang, D.; Shao, Y.; Liu, A.; Liu, X.; Ma, J. Transcriptome analysis of osmotic-responsive genes in ABA-dependent and -independent pathways in wheat (Triticum aestivum L.) roots. Peer J. 2019, 7, e6519. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zheng, Q.; Shen, W.; Cram, D.; Fowler, D.B.; Wei, Y.; Zoua, J. Understanding the Biochemical Basis of Temperature-Induced Lipid Pathway Adjustments in Plants. Plant Cell 2015, 27, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sun, Q.P.; Zhang, D.F.; Wang, T.Y.; Pan, J.B. Identification of 7 stress-related NAC transcription factor members in maize (Zea mays L.) and characterization of the expression pattern of these genes. Biochem. Biophys. Res. Commun. 2015, 462, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Yan, X.; Li, J.; Yang, Q.; Jamil, W.; Teng, Y.; Bai, S. Genome wide identification and predicted functional analyses of NAC transcription factors in Asian pears. BMC Plant Biol. 2018, 18, 214. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, Y.; Deng, X.; Liu, D.; Liu, Y.; Hu, Y.; Yan, Y. Genome-wide identification and expression analysis of expansin gene family in common wheat (Triticum aestivum L.). BMC Genom. 2019, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Guérin, C.; Roche, J.; Allard, V.; Ravel, C.; Mouzeyar, S.; Bouzidi, M.F. Genome-wide analysis, expansion and expression of the NAC family under drought and heat stresses in bread wheat (T. aestivum L.). PLoS ONE 2019, 14, e0213390. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Langley, C.H. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 1993, 260, 91–95. [Google Scholar] [CrossRef]
- Xionglei, H.; Jianzhi, Z. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 2005, 169, 1157–1164. [Google Scholar]
- He, X.; Qu, B.; Li, W.; Zhao, X.; Teng, W.; Ma, W.; Ren, Y.; Li, B.; Li, Z.; Tong, Y. The nitrate-inducible nac transcription factor TaNAC2-5a controls nitrate response and increases wheat yield. Plant Physiol. 2015, 169, 1991–2005. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, Y.; Gao, Y.; Zhou, Y.; Chen, M.; Xu, Z.; Guo, C.; Ma, Y. TaNAC48 positively regulates drought tolerance and ABA responses in wheat (Triticum aestivum L.). Crop. J. 2020. [Google Scholar] [CrossRef]
- Sanjari, S.; Shirzadian-Khorramabad, R.; Shobbar, Z.-S.; Shahbazi, M. Systematic analysis of NAC transcription factors’ gene family and identification of post-flowering drought stress responsive members in sorghum. Plant Cell Rep. 2019, 38, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yuan, Z.; Zhang, P.; Liu, Z.; Wang, T.; Wei, L. Genome-wide analysis of NAC transcription factor family in maize under drought stress and rewatering. Physiol. Mol. Biol. Plants 2020, 26, 705–717. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Q.; Shi, Y.; Sun, Z.; Zhang, Q.; Hou, S.; Wu, R.; Jiang, L.; Zhao, X.; Guo, Y. Genome-Wide Analysis of the NAC Domain Transcription Factor Gene Family in Theobroma cacao. Genes 2019, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Ali, M.; Feng, X.; Li, X. The essence of NAC gene family to the cultivation of drought-resistant soybean (Glycine max L. Merr.) cultivars. BMC Plant Biol. 2017, 17, 55. [Google Scholar] [CrossRef]
- Hu, R.; Qi, G.; Kong, Y.; Kong, D.; Gao, Q.; Zhou, G. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol. 2010, 10, 145. [Google Scholar] [CrossRef]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Meyer, T.; Muller, R.; Ploetz, L.; et al. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucleic Acids Res. 2007, 36 (Suppl. S1), D1009–D1014. [Google Scholar] [CrossRef]
- Luo, M.C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 23, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, L.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Suppl. S2), W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.; Shen, H. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Peer, Y.V.D.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Yuan, M.; Sun, B.; Zhang, D.; Zhang, J.; Li, C.; Shao, Y.; Liu, W.; Jiang, L. Evolutionary Divergence and Biased Expression of NAC Transcription Factors in Hexaploid Bread Wheat (Triticum aestivum L.). Plants 2021, 10, 382. https://doi.org/10.3390/plants10020382
Ma J, Yuan M, Sun B, Zhang D, Zhang J, Li C, Shao Y, Liu W, Jiang L. Evolutionary Divergence and Biased Expression of NAC Transcription Factors in Hexaploid Bread Wheat (Triticum aestivum L.). Plants. 2021; 10(2):382. https://doi.org/10.3390/plants10020382
Chicago/Turabian StyleMa, Jianhui, Meng Yuan, Bo Sun, Daijing Zhang, Jie Zhang, Chunxi Li, Yun Shao, Wei Liu, and Lina Jiang. 2021. "Evolutionary Divergence and Biased Expression of NAC Transcription Factors in Hexaploid Bread Wheat (Triticum aestivum L.)" Plants 10, no. 2: 382. https://doi.org/10.3390/plants10020382
APA StyleMa, J., Yuan, M., Sun, B., Zhang, D., Zhang, J., Li, C., Shao, Y., Liu, W., & Jiang, L. (2021). Evolutionary Divergence and Biased Expression of NAC Transcription Factors in Hexaploid Bread Wheat (Triticum aestivum L.). Plants, 10(2), 382. https://doi.org/10.3390/plants10020382