
SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Brassica napus

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
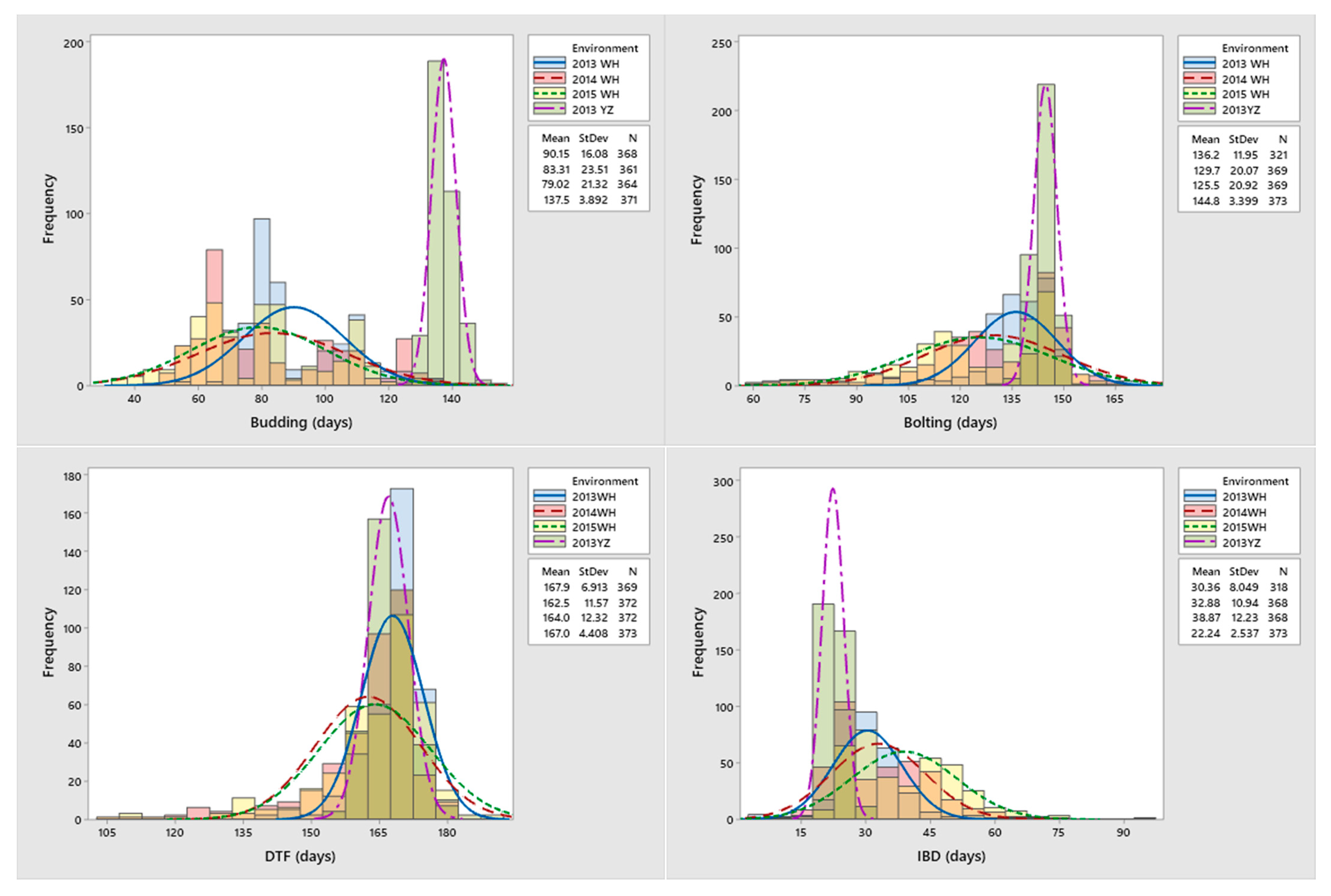
2.1. Phenotypic Variation and Correlations of Measured Traits Related to FT of B. napus
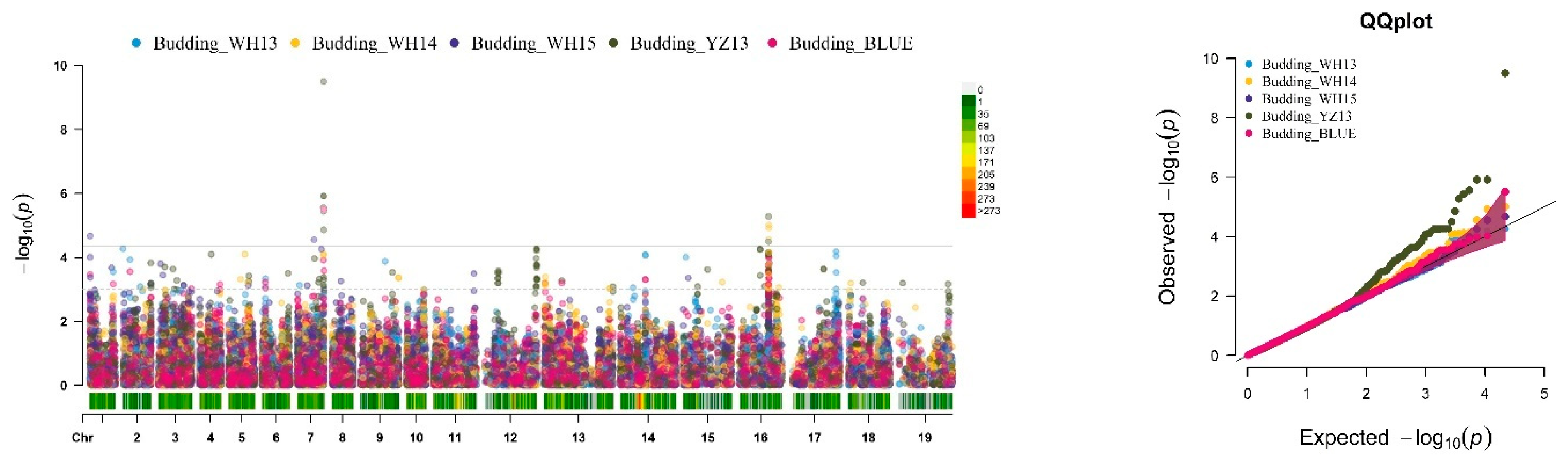
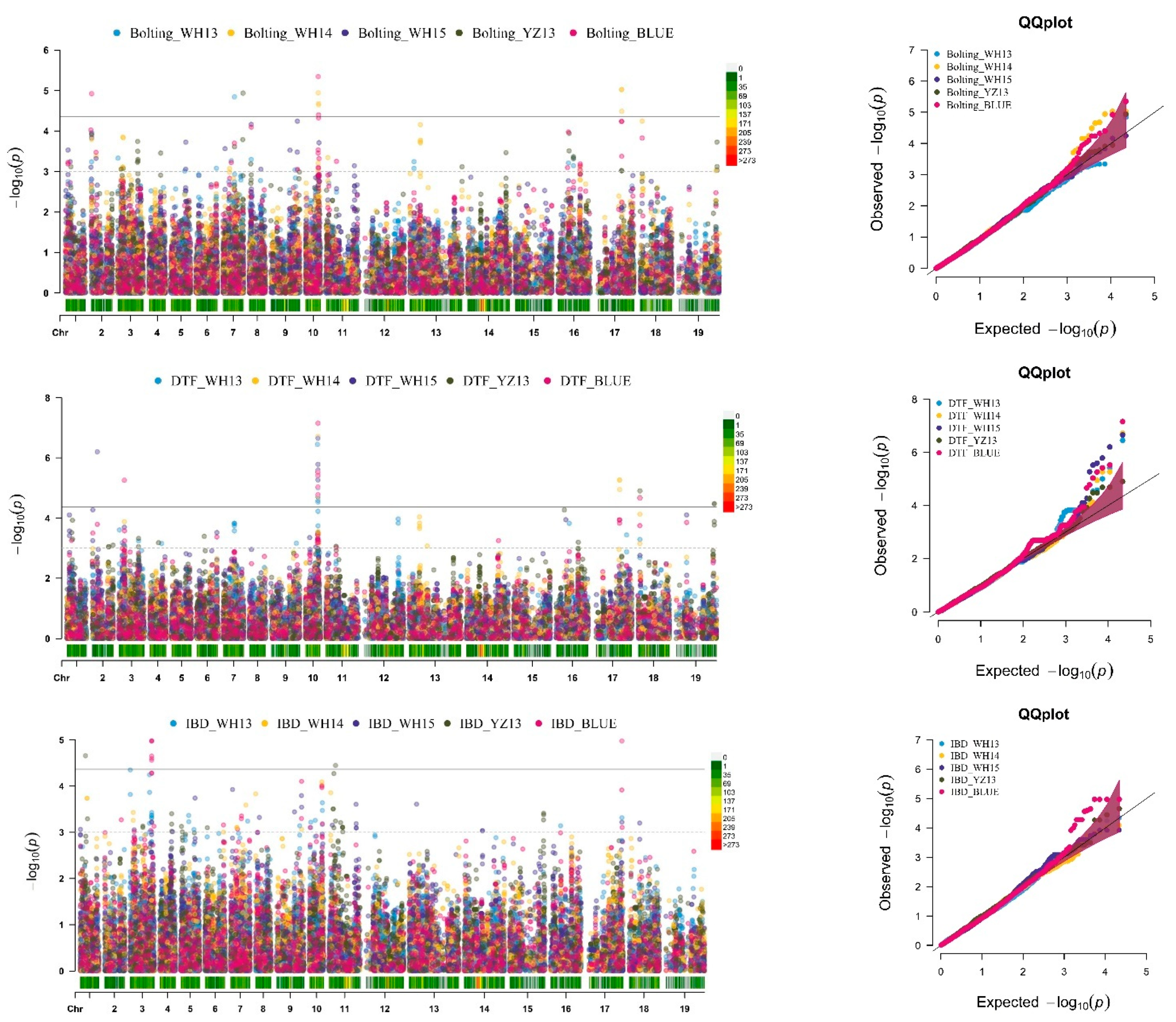
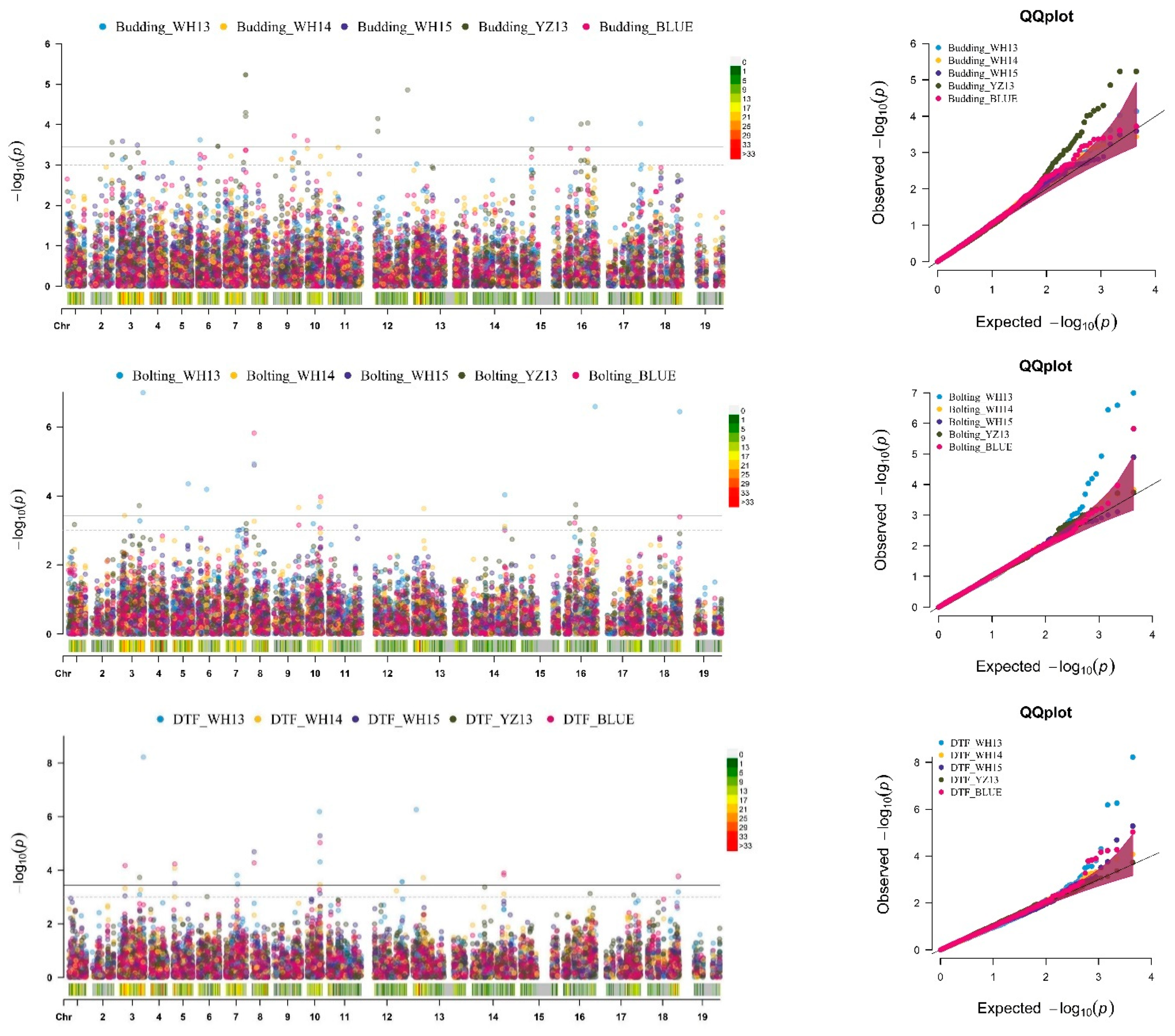
2.2. SNP-GWAS Uncovers the Candidate QTLs Targeting Traits Related to FT in B. napus
2.3. Hap-GWAS Explored QTLs Targeting Traits Related to FT in B. napus
Hap-GWAS Revealed QTL Hotspots in B. napus
2.4. Integration of SNP and Hap-GWAS with the Co-Localization of Traits Related to FT
2.4.1. GWAS Identified Important Known Genomic Regions Controlling FT in B. napus
2.4.2. Identification of Novel Genomic Regions Targeting Traits Related to FT in B. napus
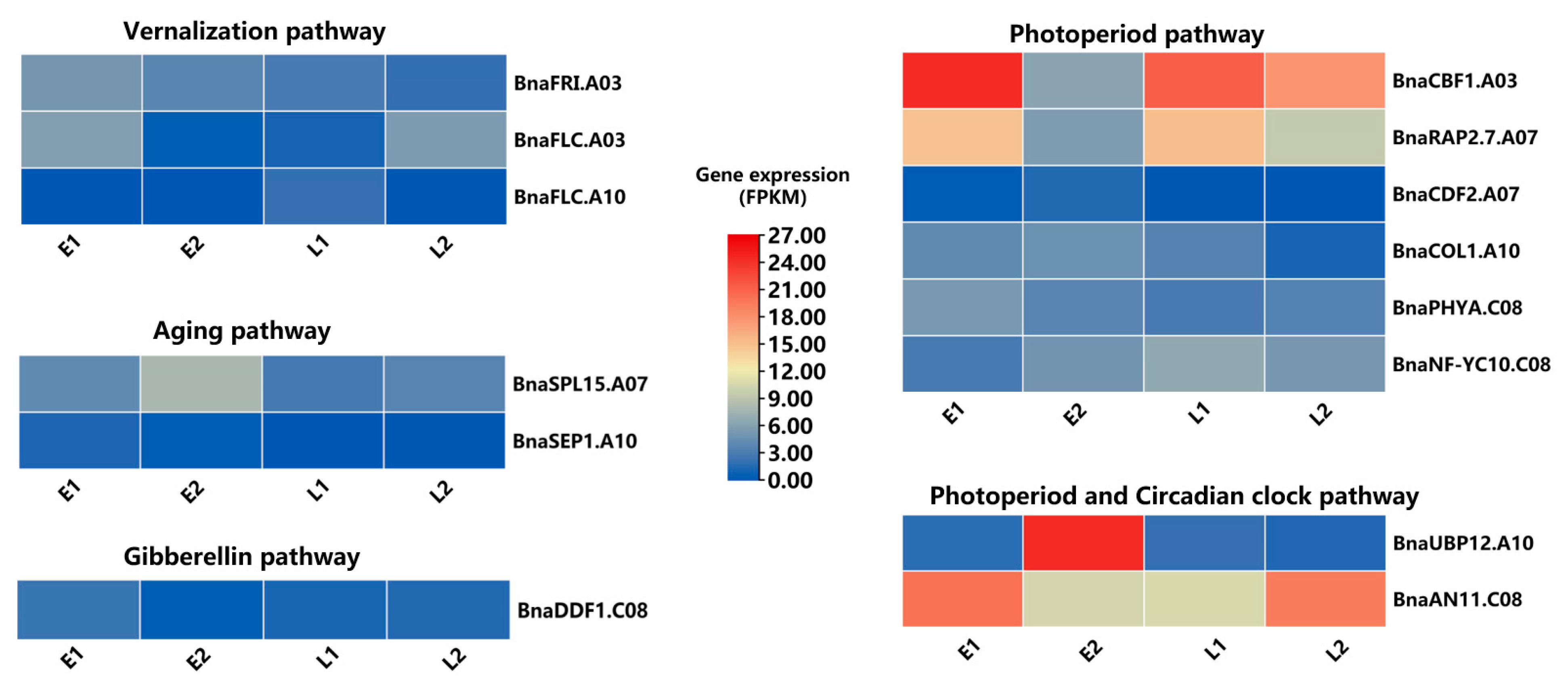
2.5. Transcriptomics Analysis
Identification of DEGs Related to Flowering Time
2.6. Prioritization of Potential Candidate Genes for FT by Integrating SNP and Hap-GWAS and Co-Localized Traits with DEGs
3. Discussion
3.1. Importance of Diverse Germplasm for Agronomic Traits Related to FT
3.2. The Implication of GWAS in Genomic-Assisted Breeding of B. napus
3.3. SNP and Hap-GWAS Co-Localized QTLs Suggested a High Degree of Pleiotropy in Controlling Traits Related to FT in Rapeseed
3.4. Comparison of SNP and Hap-GWAS Approaches to Identify QTLs Targeting Traits Related to FT in B. napus
3.5. Prioritizing the Candidate Genes to Expose the Regulatory Network of FT
4. Materials and Methods
4.1. Plant Materials and Field Experiments
4.2. Evaluation and Statistical Analysis of Phenotypes
4.3. SNP Genotyping and Quality Control
4.4. Genome-Wide Association Analysis
4.4.1. SNP-Based GWAS
4.4.2. Haplotype Based GWAS
4.5. Comparison of QTLs Targeting Flowering Time in the Present Study with Previously Reported Study
4.6. RNA-Seq/Transcriptomics Analysis
4.7. Identification of Candidate Genes and Analysis of Their Expression Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Nagaharu, U. Genome Analysis in Brassica with Special Reference to the Experimental Formation of B. Napus and Peculiar Mode of Fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T. Breeding and Utilization of Rapeseed Hybrid; Hubei Science Technology Press: Hubei, China, 2000; pp. 167–169. [Google Scholar]
- Liu, H. Genetics and Breeding in Rapeseed; Chinese Agricultural Universitatis: Beijing, China, 2000; pp. 144–177. [Google Scholar]
- Gaudinier, A.; Blackman, B.K. Evolutionary processes from the perspective of flowering time diversity. New Phytol. 2020, 225, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of Flowering in Arabidopsis. Cell 2010, 141, 550.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein Robert, J.; Zeiss, C.; Chew Emily, Y.; Tsai, J.-Y.; Sackler Richard, S.; Haynes, C.; Henning Alice, K.; SanGiovanni John, P.; Mane Shrikant, M.; Mayne Susan, T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Dreisigacker, S.; Lopes, M.; Chavez, P.; Reynolds, M.P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 2015, 128, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, K.; Zhang, Z.; Guan, C.; Chen, S.; Hua, W.; Li, J.; Wen, J.; Yi, B.; Shen, J.; et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 2016, 23, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Jian, H.; Zhang, A.; Ma, J.; Wang, T.; Yang, B.; Shuang, L.S.; Liu, M.; Li, J.; Xu, X.; Paterson, A.H.; et al. Joint QTL mapping and transcriptome sequencing analysis reveal candidate flowering time genes in Brassica napus L. BMC Genom. 2019, 20, 21. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Coombes, N.; Song, J.; Prangnell, R.; Bandaranayake, C.; Tahira, R.; Sundaramoorthi, V.; Killian, A.; Meng, J.; et al. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ. 2016, 39, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Han, D.; Mason, A.S.; Zhou, C.; Zheng, W.; Li, Y.; Wu, C.; Fu, D.; Huang, Y. Earliness traits in rapeseed (Brassica napus): SNP loci and candidate genes identified by genome-wide association analysis. DNA Res. 2018, 25, 229–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wei, L.; Wang, J.; Xie, L.; Li, Y.Y.; Ran, S.; Ren, L.; Lu, K.; Li, J.; Timko, M.P.; et al. Integrating GWAS, linkage mapping and gene expression analyses reveals the genetic control of growth period traits in rapeseed (Brassica napus L.). Biotechnol. Biofuels 2020, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, W.; Li, D.; Chao, H.; Zhao, X.; Ta, N.; Li, Y.; Guan, Z.; Guo, L.; Zhang, L.; et al. Genetic dissection of the mechanism of flowering time based on an environmentally stable and specific QTL in Brassica napus. Plant Sci. 2018, 277, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Abed, A.; Belzile, F. Comparing Single-SNP, Multi-SNP, and Haplotype-Based Approaches in Association Studies for Major Traits in Barley. Plant Genome 2019, 12, 190036. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Jiang, Y.; Zhao, Y.; Schulthess, A.W.; Reif, J.C. Haplotype-based genome-wide association increases the predictability of leaf rust (Puccinia triticina) resistance in wheat. J. Exp. Bot. 2020, 71, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, D.; Rosyara, U.; Mondal, S.; Singh, R.; Poland, J.; Dreisigacker, S. Incorporating Genome-Wide Association Mapping Results Into Genomic Prediction Models for Grain Yield and Yield Stability in CIMMYT Spring Bread Wheat. Front. Plant Sci. 2020, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Mokry, F.B.; Buzanskas, M.E.; de Alvarenga Mudadu, M.; do Amaral Grossi, D.; Higa, R.H.; Ventura, R.V.; de Lima, A.O.; Sargolzaei, M.; Conceição Meirelles, S.L.; Schenkel, F.S.; et al. Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genom. 2014, 15 (Suppl. 7), S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachi, B.; Faure, N.; Bergelson, J.; Cuguen, J.; Roux, F. Genome-wide association mapping of flowering time in Arabidopsis thaliana in nature: Genetics for underlying components and reaction norms across two successive years. Acta Bot. Gall. 2013, 160, 205–219. [Google Scholar] [CrossRef]
- Sukumaran, S.; Lopes, M.; Dreisigacker, S.; Reynolds, M. Genetic analysis of multi-environmental spring wheat trials identifies genomic regions for locus-specific trade-offs for grain weight and grain number. Theor. Appl. Genet. 2018, 131, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Weinholdt, C.; Jedrusik, N.; Molina, C.; Zou, J.; Große, I.; Schiessl, S.; Jung, C.; Emrani, N. Whole-transcriptome analysis reveals genetic factors underlying flowering time regulation in rapeseed (Brassica napus L.). Plant Cell Environ. 2018, 41, 1935–1947. [Google Scholar] [CrossRef]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Xiang, Y.; Xu, E.; Ge, X.; Li, Z. Major Co-localized QTL for Plant Height, Branch Initiation Height, Stem Diameter, and Flowering Time in an Alien Introgression Derived Brassica napus DH Population. Front. Plant Sci. 2018, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Wang, X.; Li, W.; Sun, L.; Peng, Q.; Chen, F.; Zhang, W.; Guan, R.; Zhang, J. Identification and physical mapping of QTLs associated with flowering time in Brassica napus L. Euphytica 2019, 215, 152. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Lu, Y.; Shah, T.; Hao, Z.; Taba, S.; Zhang, S.; Gao, S.; Liu, J.; Cao, M.; Wang, J.; Prakash, A.B.; et al. Comparative SNP and Haplotype Analysis Reveals a Higher Genetic Diversity and Rapider LD Decay in Tropical than Temperate Germplasm in Maize. PLoS ONE 2011, 6, e24861. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Qiao, J.; Yan, G.; Li, J.; Li, H.; Wu, X. Association Mapping of Flowering Time QTLs and Insight into Their Contributions to Rapeseed Growth Habits. Front. Plant Sci. 2016, 7, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Cui, Y.; He, Y.; Xiong, Q.; Qian, L.; Tong, C.; Lu, G.; Ding, Y.; Li, J.; Jung, C.; et al. A genome-wide survey with different rapeseed ecotypes uncovers footprints of domestication and breeding. J. Exp. Bot. 2017, 68, 4791–4801. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-Genome Resequencing of a Worldwide Collection of Rapeseed Accessions Reveals the Genetic Basis of Ecotype Divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, P.; Zhao, Q.; Cai, G.; Hu, Y.; Xiang, Y.; Yang, Q.; Wang, Y.; Zhou, Y. Co-location of QTL for Sclerotinia stem rot resistance and flowering time in Brassica napus. Crop J. 2019, 7, 227–237. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, B.; Ma, N.; Liu, X.; Qin, M.; Zhang, Y.; Wang, K.; Guo, N.; Zuo, K.; Liu, X.; et al. Quantitative Trait Locus Mapping and Identification of Candidate Genes Controlling Flowering Time in Brassica napus L. Front. Plant Sci. 2021, 11, 2321. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C Encodes a Novel MADS Domain Protein That Acts as a Repressor of Flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Robert, L.S.; Robson, F.; Sharpe, A.; Lydiate, D.; Coupland, G. Conserved structure and function of the Arabidopsis flowering time gene CONSTANS in Brassica napus. Plant Mol. Biol. 1998, 37, 763–772. [Google Scholar] [CrossRef]
- An, H.; Qi, X.; Gaynor, M.L.; Hao, Y.; Gebken, S.C.; Mabry, M.E.; McAlvay, A.C.; Teakle, G.R.; Conant, G.C.; Barker, M.S.; et al. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nat. Commun. 2019, 10, 2878. [Google Scholar] [CrossRef] [Green Version]
- McClung, C.R. Plant Circadian Rhythms. Plant Cell 2006, 18, 792–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhu, Y.; Varshney, R.K.; Zhan, J.; Zheng, X.; Shi, J.; Wang, X.; Liu, G.; Wang, H. A systematic dissection of the mechanisms underlying the natural variation of silique number in rapeseed (Brassica napus L.) germplasm. Plant Biotechnol. J. 2020, 18, 568–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merikangas, K.R.; Risch, N. Genomic Priorities and Public Health. Science 2003, 302, 599. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef]
- Li, F.; Chen, B.; Xu, K.; Wu, J.; Song, W.; Bancroft, I.; Harper, A.L.; Trick, M.; Liu, S.; Gao, G.; et al. Genome-Wide Association Study Dissects the Genetic Architecture of Seed Weight and Seed Quality in Rapeseed (Brassica napus L.). DNA Res. 2014, 21, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Chen, B.; Xu, K.; Gao, G.; Yan, G.; Qiao, J.; Li, J.; Li, H.; Li, L.; Xiao, X.; et al. A genome-wide association study of plant height and primary branch number in rapeseed (Brassica napus). Plant Sci. 2016, 242, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Xiao, Y.; Yang, W.; Ye, W.; Wang, B.; Younas, M.; Wu, J.; Liu, K. Association mapping of six yield-related traits in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2014, 127, 85–96. [Google Scholar] [CrossRef]
- Wei, L.; Jian, H.; Lu, K.; Filardo, F.; Yin, N.; Liu, L.; Qu, C.; Li, W.; Du, H.; Li, J. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 2016, 14, 1368–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. 2013, 29, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kenney-Hunt, J.P.; Pavlicev, M.; Peck, J.R.; Waxman, D.; Cheverud, J.M. Pleiotropic scaling of gene effects and the ‘cost of complexity’. Nature 2008, 452, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jeong, K.; Davis, J.T.; Kim, S.; Lee, S.; Michelmore, R.W.; Kim, S.; Maloof, J.N. Integrated QTL and eQTL Mapping Provides Insights and Candidate Genes for Fatty Acid Composition, Flowering Time, and Growth Traits in a F2 Population of a Novel Synthetic Allopolyploid Brassica napus. Front. Plant Sci. 2018, 9, 1632. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Auge, G.A.; Penfield, S.; Donohue, K. Pleiotropy in developmental regulation by flowering-pathway genes: Is it an evolutionary constraint? New Phytol. 2019, 224, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, G.C.K.; Barua, D.; Kramer, E.M.; Amasino, R.M.; Donohue, K. Major flowering time gene, FLOWERING LOCUS C, regulates seed germination in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2009, 106, 11661. [Google Scholar] [CrossRef] [Green Version]
- Willmann, M.R.; Poethig, R.S. The effect of the floral repressor FLC on the timing and progression of vegetative phase change in Arabidopsis. Development 2011, 138, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, A.J.; Hamblin, M.T.; Jannink, J.-L. Performance of single nucleotide polymorphisms versus haplotypes for genome-wide association analysis in barley. PLoS ONE 2010, 5, e14079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Soto, R.I.; Mora, F.; de Oliveira, M.A.R.; Higashi, W.; Scapim, C.A.; Schuster, I. A Genome-Wide Association Study for Agronomic Traits in Soybean Using SNP Markers and SNP-Based Haplotype Analysis. PLoS ONE 2017, 12, e0171105. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, A.; Haile, J.K.; Cory, A.T.; Clarke, F.R.; Clarke, J.M.; Knox, R.E.; Pozniak, C.J. Single Marker and Haplotype-Based Association Analysis of Semolina and Pasta Colour in Elite Durum Wheat Breeding Lines Using a High-Density Consensus Map. PLoS ONE 2017, 12, e0170941. [Google Scholar]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [Green Version]
- Körber, N.; Bus, A.; Li, J.; Parkin, I.A.P.; Wittkop, B.; Snowdon, R.J.; Stich, B. Agronomic and Seed Quality Traits Dissected by Genome-Wide Association Mapping in Brassica napus. Front. Plant Sci. 2016, 7, 386. [Google Scholar] [CrossRef]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Notaguchi, M.; Abe, M.; Kimura, T.; Daimon, Y.; Kobayashi, T.; Yamaguchi, A.; Tomita, Y.; Dohi, K.; Mori, M.; Araki, T. Long-distance, graft-transmissible action of Arabidopsis FLOWERING LOCUS T protein to promote flowering. Plant Cell Physiol. 2008, 49, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Dong, F.; Cai, J.; Xin, Q.; Fang, C.; Liu, L.; Wan, L.; Yang, G.; Hong, D. A 2.833-kb Insertion in BnFLC.A2 and Its Homeologous Exchange with BnFLC.C2 during Breeding Selection Generated Early-Flowering Rapeseed. Mol. Plant 2018, 11, 222–225. [Google Scholar] [CrossRef]
- Yin, S.; Wan, M.; Guo, C.; Wang, B.; Li, H.; Li, G.; Tian, Y.; Ge, X.; King, G.J.; Liu, K.; et al. Transposon insertions within alleles of BnaFLC.A10 and BnaFLC.A2 are associated with seasonal crop type in rapeseed. J. Exp. Bot. 2020, 71, 4729–4741. [Google Scholar] [CrossRef]
- Wang, N.; Qian, W.; Suppanz, I.; Wei, L.; Mao, B.; Long, Y.; Meng, J.; Müller, A.E.; Jung, C. Flowering time variation in oilseed rape (Brassica napus L.) is associated with allelic variation in the FRIGIDA homologue BnaA.FRI.a. J. Exp. Bot. 2011, 62, 5641–5658. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-H.; Mitina, I.; Tamada, Y.; Hyun, Y.; Choi, Y.; Amasino, R.M.; Noh, B.; Noh, Y.-S. Growth habit determination by the balance of histone methylation activities in Arabidopsis. EMBO J. 2010, 29, 3208–3215. [Google Scholar] [CrossRef] [Green Version]
- Tadege, M.; Sheldon, C.C.; Helliwell, C.A.; Stoutjesdijk, P.; Dennis, E.S.; Peacock, W.J. Control of flowering time by FLC orthologues in Brassica napus. Plant J. 2001, 28, 545–553. [Google Scholar] [CrossRef]
- Helliwell, C.A.; Wood, C.C.; Robertson, M.; James Peacock, W.; Dennis, E.S. The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 2006, 46, 183–192. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. Loss of FLOWERING LOCUS C Activity Eliminates the Late-Flowering Phenotype of FRIGIDA and Autonomous Pathway Mutations but Not Responsiveness to Vernalization. Plant Cell 2001, 13, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Long, Y.; Raman, H.; Zou, X.; Wang, J.; Dai, S.; Xiao, Q.; Li, C.; Fan, L.; Liu, B.; et al. A Tourist-like MITE insertion in the upstream region of the BnFLC.A10 gene is associated with vernalization requirement in rapeseed (Brassica napus L.). BMC Plant Biol. 2012, 12, 238. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, C.; Dean, C. The FLC Locus: A Platform for Discoveries in Epigenetics and Adaptation. Annu. Rev. Cell Dev. Biol. 2017, 33, 555–575. [Google Scholar] [CrossRef]
- Fornara, F.; Panigrahi, K.C.S.; Gissot, L.; Sauerbrunn, N.; Rühl, M.; Jarillo, J.A.; Coupland, G. Arabidopsis DOF Transcription Factors Act Redundantly to Reduce CONSTANS Expression and Are Essential for a Photoperiodic Flowering Response. Dev. Cell 2009, 17, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Guo, T.; Liu, Y.; Liu, Q.; Fang, Y. The Roles of Arabidopsis CDF2 in Transcriptional and Posttranscriptional Regulation of Primary MicroRNAs. PLoS Genet. 2015, 11, e1005598. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kiba, T.; Chua, N.-H. The Arabidopsis SPA1 gene is required for circadian clock function and photoperiodic flowering. Plant J. 2006, 46, 736–746. [Google Scholar] [CrossRef]
- Song, Y.H.; Kubota, A.; Kwon, M.S.; Covington, M.F.; Lee, N.; Taagen, E.R.; Laboy Cintrón, D.; Hwang, D.Y.; Akiyama, R.; Hodge, S.K.; et al. Molecular basis of flowering under natural long-day conditions in Arabidopsis. Nat. Plants 2018, 4, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Lenth, R.V. Least-Squares Means: The R Package lsmeans. J. Stat. Softw. 2016, 69, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dong, C.; Hu, M.; Bai, Z.; Tong, C.; Zuo, R.; Liu, Y.; Cheng, X.; Cheng, M.; Huang, J.; et al. Identification of Flower-Specific Promoters through Comparative Transcriptome Analysis in Brassica napus. Int. J. Mol. Sci. 2019, 20, 5949. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Trait | Mean (Days) | SD | CV (%) | Kurtosis | Skewness | Heritability (h2) |
|---|---|---|---|---|---|---|---|
| 2013WH | Budding | 90.15 | 16.08 | 17.84 | −0.03 | 0.81 | 0.90 |
| Bolting | 136.22 | 11.95 | 8.77 | 6.37 | −1.66 | 0.94 | |
| DTF | 167.89 | 6.91 | 4.12 | 11.09 | −2.24 | 0.85 | |
| IBD | 25.88 | 13.09 | 50.58 | 0.98 | −0.52 | 0.84 | |
| 2014WH | Budding | 83.31 | 23.51 | 28.22 | −0.62 | 0.69 | 0.93 |
| Bolting | 129.68 | 20.07 | 15.48 | 1.00 | −1.02 | 0.92 | |
| DTF | 162.49 | 11.57 | 7.12 | 4.91 | −1.97 | 0.94 | |
| IBD | 32.44 | 11.51 | 35.48 | 0.37 | 0.36 | 0.88 | |
| 2015WH | Budding | 79.02 | 21.32 | 26.98 | −0.61 | 0.39 | 0.98 |
| Bolting | 125.47 | 20.92 | 16.68 | 0.02 | −0.75 | 0.98 | |
| DTF | 164.00 | 12.32 | 7.51 | 4.67 | −1.79 | 0.97 | |
| IBD | 38.35 | 12.94 | 33.75 | 0.80 | 0.37 | 0.96 | |
| 2013YZ | Budding | 137.50 | 3.89 | 2.83 | 1.57 | 0.84 | 0.57 |
| Bolting | 144.79 | 3.40 | 2.35 | 0.90 | −0.43 | 0.53 | |
| DTF | 167.03 | 4.41 | 2.64 | 0.12 | 0.14 | 0.64 | |
| IBD | 22.24 | 2.54 | 11.41 | 4.39 | −0.56 | 0.73 |
| Integrated/ Co-localized region | B napus gene ID | Gene Name | AT Ortholog | Gene Annotation | SNP QTL | Hap-QTL | Traits | Environment | SE1vSL1 | SE1vSL2 | SE2vSL1 | SE2vSL2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FT.A03.1 | BnaA03g13320D | FRI | AT4G00650.1 | FRIGIDA | qFT.A03.1 | hap.qFT.A03.1 | Budding, DTF | WH15, BLUE | 0.6464 | 1.3505 | 0.2236 | 0.9277 |
| BnaA03g13620D | CBF1 | AT4G25490.1 | C-repeat/DRE binding factor 1 | 0.0628 | 0.5349 | −1.8762 | −1.4040 | |||||
| BnaA03g13630D | FLC | AT5G10140.1 | FLOWERING LOCUS C | 2.3706 | 0.0129 | −0.6333 | −2.9910 | |||||
| FT.A07.1 | BnaA07g13990D | RAP2.7 | AT2G28550.3 | Related to AP2.7 | qFT.A07.1 | hap.qFT.A07.1 | Budding, Bolting, IBD | WH13, WH15 | −0.0614 | 0.4684 | −1.4487 | −0.9188 |
| BnaA07g14740D | CDF2 | AT5G39660.2 | Cycling DOF factor 2 | 4.8967 | 5.7036 | 5.9862 | 6.7932 | |||||
| BnaA07g17550D | SPL15 | AT3G57920.1 | Squamosa promoter binding protein-like 15 | 0.3184 | −0.1571 | 1.3920 | 0.9165 | |||||
| FT.A10 | BnaA10g18420D | COL1 | AT5G15850.1 | CONSTANS-like 1 | qFT.A10 | hap.qFT.A10.3 | Bolting, DTF, IBD | WH13, WH14, WH15, YZ13, BLUE | −0.0295 | 1.7775 | 0.5624 | 2.3694 |
| BnaA10g18480D | (SEP1) | AT5G15800.1 | SEPALLATA1 | 5.7243 | 5.5249 | 4.5259 | 4.3265 | |||||
| BnaA10g22080D | FLC | AT5G10140.1 | FLOWERING LOCUS C | −7.3033 | −3.2291 | −6.9579 | −2.8836 | |||||
| BnaA10g24300D | UBP12 | AT5G06600.1 | Ubiquitin-specific protease 12 | −0.5266 | 0.3302 | 3.2339 | 4.0907 | |||||
| FT.C08 | BnaC08g40840D | ATAN11 | AT1G12910.1 | ANTHOCYANIN11 | hap.qFT.C08 | Bolting, DTF | WH13, WH15, BLUE | 1.0874 | −0.0768 | −0.0832 | −1.2475 | |
| BnaC08g41070D | DDF1 | AT1G12610.1 | DWARF AND DELAYED FLOWERING 1 | 0.9130 | 0.6218 | −1.3937 | −1.6849 | |||||
| BnaC08g42660D | PHYA | AT1G09570.1 | Phytochrome A | 1.0181 | 0.7956 | 0.4025 | 0.1801 | |||||
| BnaC08g43430D | NF-YC10 | AT1G07980.1 | Nuclear factor Y, subunit C10 | −1.2312 | −0.7080 | −0.3565 | 0.1666 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helal, M.; Gill, R.A.; Tang, M.; Yang, L.; Hu, M.; Yang, L.; Xie, M.; Zhao, C.; Cheng, X.; Zhang, Y.; et al. SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Brassica napus. Plants 2021, 10, 2475. https://doi.org/10.3390/plants10112475
Helal M, Gill RA, Tang M, Yang L, Hu M, Yang L, Xie M, Zhao C, Cheng X, Zhang Y, et al. SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Brassica napus. Plants. 2021; 10(11):2475. https://doi.org/10.3390/plants10112475
Chicago/Turabian StyleHelal, MMU, Rafaqat Ali Gill, Minqiang Tang, Li Yang, Ming Hu, Lingli Yang, Meili Xie, Chuanji Zhao, Xiaohui Cheng, Yuanyuan Zhang, and et al. 2021. "SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Brassica napus" Plants 10, no. 11: 2475. https://doi.org/10.3390/plants10112475
APA StyleHelal, M., Gill, R. A., Tang, M., Yang, L., Hu, M., Yang, L., Xie, M., Zhao, C., Cheng, X., Zhang, Y., Zhang, X., & Liu, S. (2021). SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Brassica napus. Plants, 10(11), 2475. https://doi.org/10.3390/plants10112475