
An Evolutionary Perspective on Hox Binding Site Preferences in Two Different Tissues


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
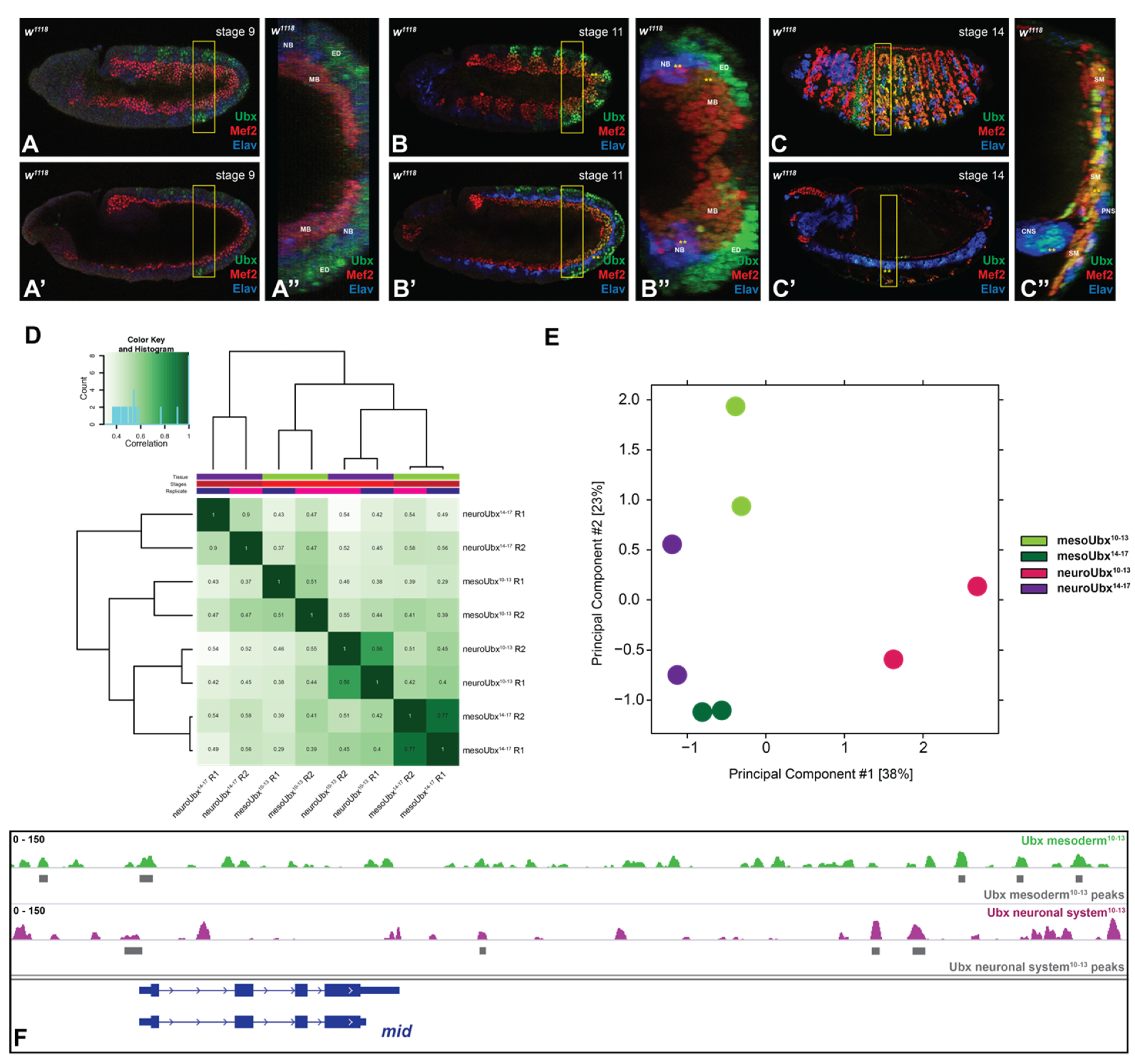
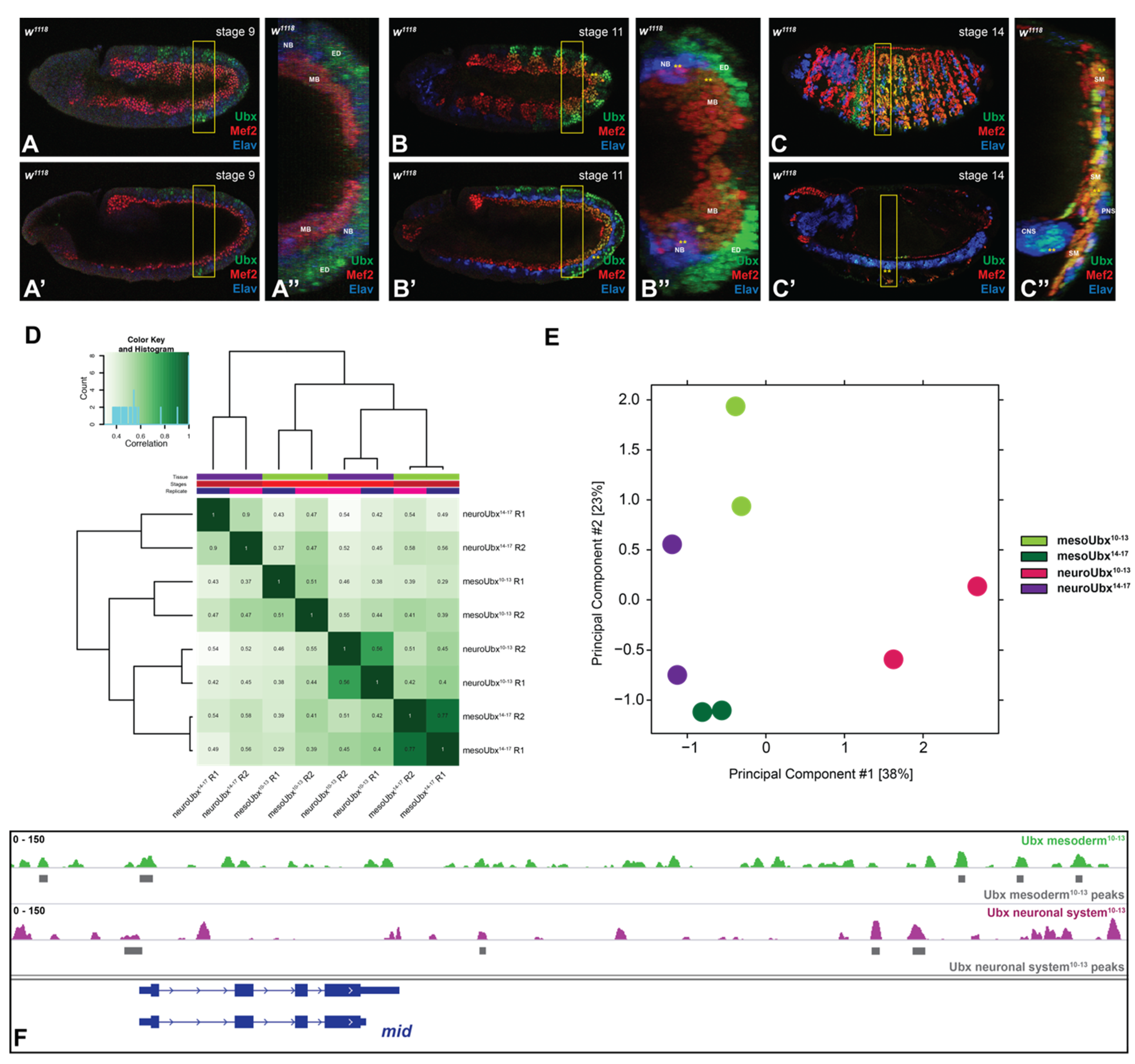
2.1. Ubx Chromatin Interactions Are Highly Specific in Two Different Tissues
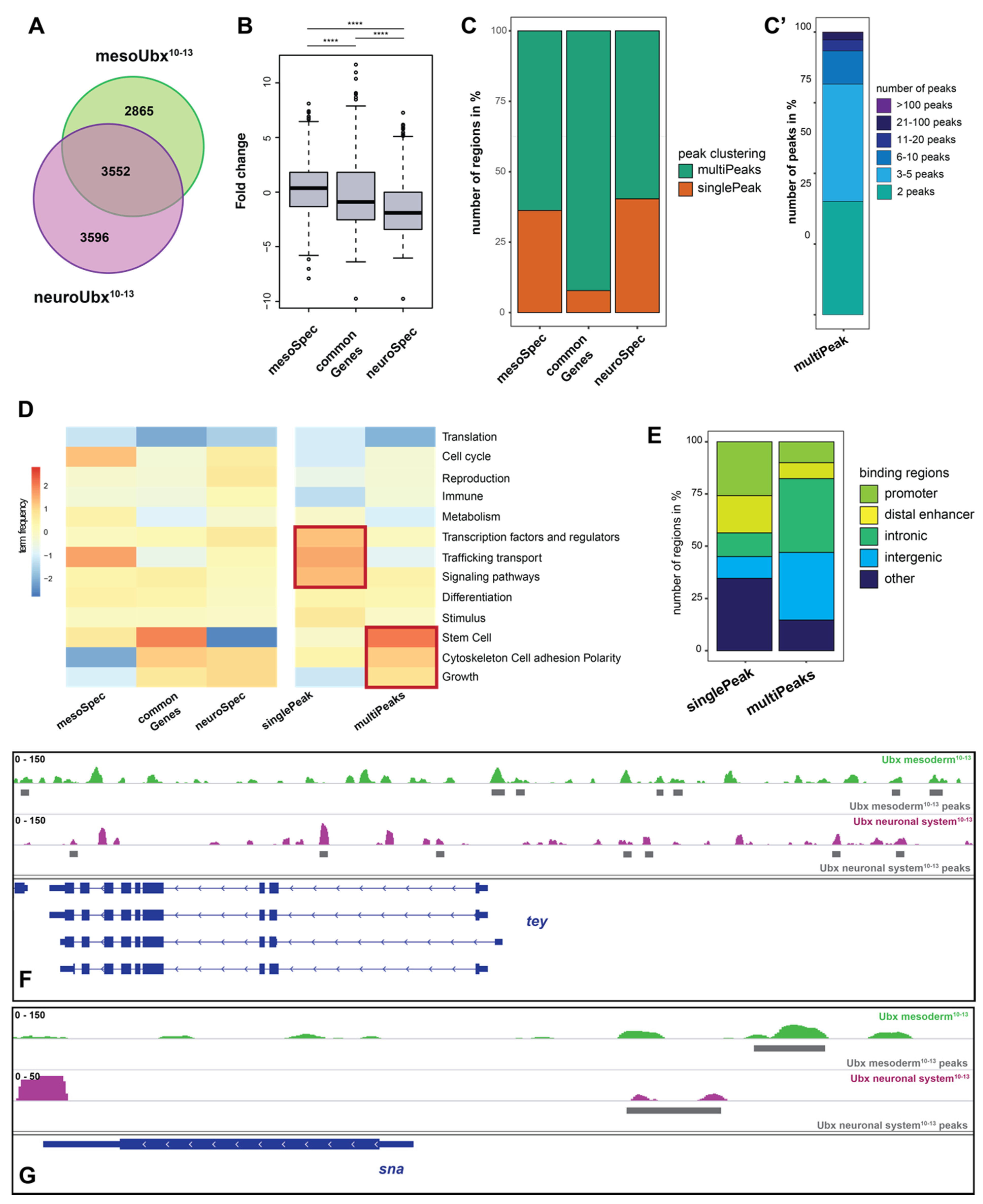
2.2. Ubx Mostly Interacts with Multiple Chromatin Sites in the Different Tissues
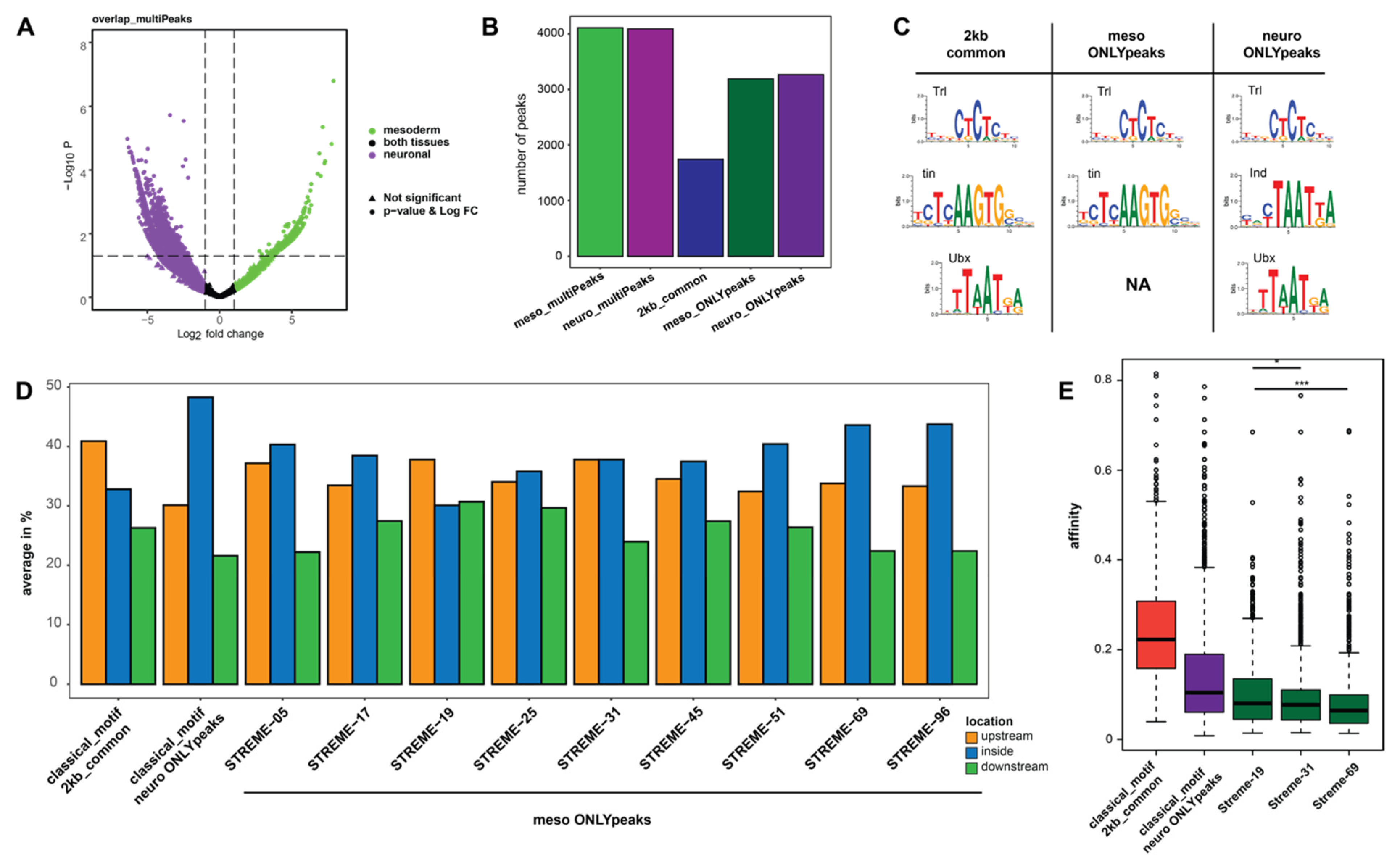
2.3. Novel Sequence Patterns Resembling Hox Motifs Are Enriched at Ubx Chromatin Sites in the Mesoderm
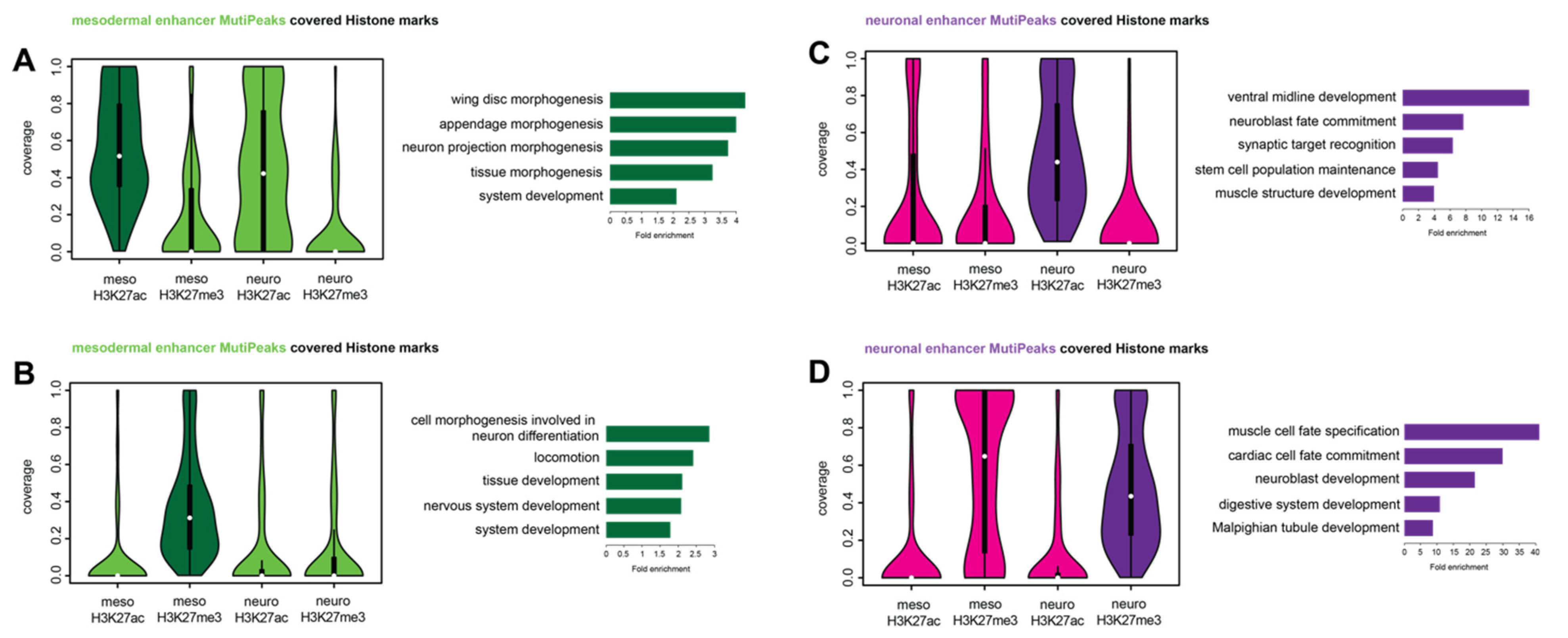
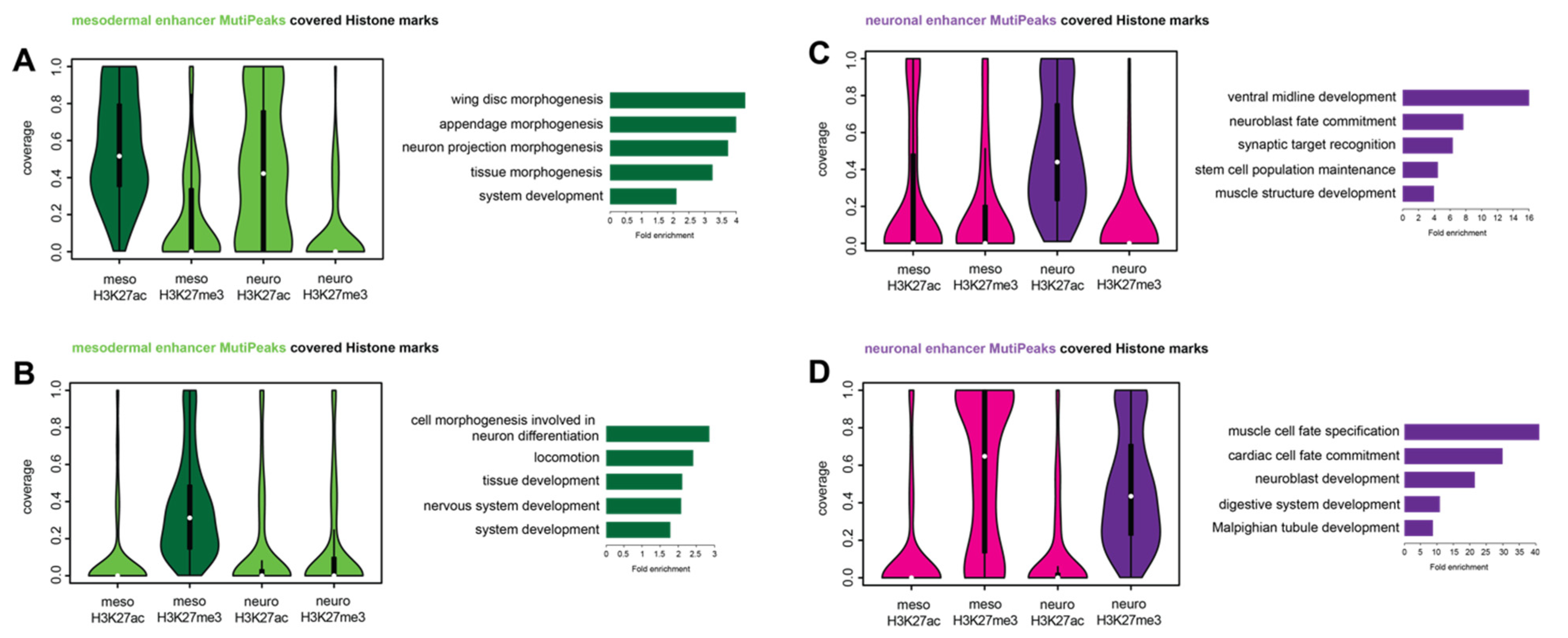
2.4. Tissue-Specific Ubx Sites Show Differences in Histone Mark Distributions
3. Discussion
4. Materials and Methods
4.1. Fly Stocks and Antibody Staining
4.2. Bioinformatic Analysis and Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandmann, T.; Girardot, C.; Brehme, M.; Tongprasit, W.; Stolc, V.; Furlong, E.E. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev. 2007, 21, 436–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, T.L.; Rebay, I. Master regulators in development: Views from the Drosophila retinal determination and mammalian pluripotency gene networks. Dev. Biol. 2017, 421, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr. Biol. 2016, 26, 2101–2113. [Google Scholar] [CrossRef] [Green Version]
- Domsch, K.; Carnesecchi, J.; Disela, V.; Friedrich, J.; Trost, N.; Ermakova, O.; Polychronidou, M.; Lohmann, I. The Hox transcription factor Ubx stabilizes lineage commitment by suppressing cellular plasticity in Drosophila. Elife 2019, 8, e42675. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, S.; Westholm, J.O.; Dai, Q.; Matsuda, R.; Hosono, C.; Bray, S.; Lai, E.C.; Samakovlis, C. Genome-wide identification of Grainy head targets in Drosophila reveals regulatory interactions with the POU domain transcription factor Vvl. Development 2017, 144, 3145–3155. [Google Scholar] [CrossRef] [Green Version]
- Sandmann, T.; Jensen, L.J.; Jakobsen, J.S.; Karzynski, M.M.; Eichenlaub, M.P.; Bork, P.; Furlong, E.E. A temporal map of transcription factor activity: Mef2 directly regulates target genes at all stages of muscle development. Dev. Cell 2006, 10, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Stojnic, R.; Adryan, B.; Ozdemir, A.; Stathopoulos, A.; Frasch, M. Genome-wide screens for in vivo Tinman binding sites identify cardiac enhancers with diverse functional architectures. PLoS Genet. 2013, 9, e1003195. [Google Scholar] [CrossRef] [Green Version]
- Akam, M. The molecular basis for metameric pattern in the Drosophila embryo. Development 1987, 101, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.P.; Carroll, S.B. The segmentation and homeotic gene network in early Drosophila development. Cell 1987, 51, 689–698. [Google Scholar] [CrossRef]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.; Papalopulu, N.; Krumlauf, R. The murine and Drosophila homeobox gene complexes have common features of organization and expression. Cell 1989, 57, 367–378. [Google Scholar] [CrossRef]
- Berger, M.F.; Badis, G.; Gehrke, A.R.; Talukder, S.; Philippakis, A.A.; Pena-Castillo, L.; Alleyne, T.M.; Mnaimneh, S.; Botvinnik, O.B.; Chan, E.T.; et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 2008, 133, 1266–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, R.S.; Lelli, K.M.; Joshi, R. Hox specificity unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 2009, 88, 63–101. [Google Scholar] [PubMed] [Green Version]
- Noyes, M.B.; Christensen, R.G.; Wakabayashi, A.; Stormo, G.D.; Brodsky, M.H.; Wolfe, S.A. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 2008, 133, 1277–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.; Riley, T.; Liu, P.; Abe, N.; Gomez-Alcala, P.; Dror, I.; Zhou, T.; Rohs, R.; Honig, B.; Bussemaker, H.J.; et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 2011, 147, 1270–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocker, J.; Abe, N.; Rinaldi, L.; McGregor, A.P.; Frankel, N.; Wang, S.; Alsawadi, A.; Valenti, P.; Plaza, S.; Payre, F.; et al. Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 2015, 160, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Higueras, C.; Rastogi, C.; Voutev, R.; Bussemaker, H.J.; Mann, R.S.; Hombria, J.C. In Vivo Hox binding specificity revealed by systematic changes to a single cis regulatory module. Nat. Commun. 2019, 10, 3597. [Google Scholar] [CrossRef] [Green Version]
- Pearson, J.C.; Lemons, D.; McGinnis, W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 2005, 6, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Steiner, F.A.; Talbert, P.B.; Kasinathan, S.; Deal, R.B.; Henikoff, S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res. 2012, 22, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Trost, N.; Rempel, E.; Ermakova, O.; Tamirisa, S.; Parcalabescu, L.; Boutros, M.; Lohmann, J.U.; Lohmann, I. WEADE: A workflow for enrichment analysis and data exploration. PLoS ONE 2018, 13, e0204016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- McLeay, R.C.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinform. 2010, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nat. Cell Biol. 2010, 471, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonn, S.; Zinzen, R.; Girardot, C.; Gustafson, E.H.; Gonzalez, A.P.; Delhomme, N.; Ghavi-Helm, Y.; Wilczynski, B.; Riddell, A.; Furlong, E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nat. Cell Biol. 2010, 470, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Hueber, S.D.; Bezdan, D.; Henz, S.R.; Blank, M.; Wu, H.; Lohmann, I. Comparative analysis of Hox downstream genes in Drosophila. Development 2007, 134, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Hueber, S.D.; Lohmann, I. Shaping segments: Hox gene function in the genomic age. Bioessays 2008, 30, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Krumlauf, R. Hox Genes and the Hindbrain: A Study in Segments. Curr. Top. Dev. Biol. 2016, 116, 581–596. [Google Scholar]
- Burton, P.M. Insights from diploblasts; the evolution of mesoderm and muscle. J. Exp. Zool. B Mol. Dev. Evol. 2008, 310, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Technau, U.; Genikhovich, G. Evolution: Directives from Sea Anemone Hox Genes. Curr. Biol. 2018, 28, R1303–R1305. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Feder, M.; Levin, M.; Hall, B.K.; Yanai, I. Spatiotemporal transcriptomics reveals the evolutionary history of the endoderm germ layer. Nature 2015, 519, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Technau, U.; Scholz, C.B. Origin and evolution of endoderm and mesoderm. Int. J. Dev. Biol. 2003, 47, 531–539. [Google Scholar] [PubMed]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Program, N.C.S.; et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Del Viso, F.; Chen, C.Y.; Ikmi, A.; Kroesen, A.E.; Gibson, M.C. An axial Hox code controls tissue segmentation and body patterning in Nematostella vectensis. Science 2018, 361, 1377–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusanovich, D.A.; Reddington, J.P.; Garfield, D.A.; Daza, R.M.; Aghamirzaie, D.; Marco-Ferreres, R.; Pliner, H.A.; Christiansen, L.; Qiu, X.; Steemers, F.J.; et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 2018, 555, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Harrison, M.M.; Villalta, J.E.; Kaplan, T.; Eisen, M.B. Establishment of regions of genomic activity during the Drosophila maternal to zygotic transition. Elife 2014, 3, e03737. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Johnston, J.; Shao, W.; Meier, S.; Staber, C.; Zeitlinger, J. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. Elife 2013, 2, e00861. [Google Scholar] [CrossRef] [PubMed]
- Domsch, K.; Ezzeddine, N.; Nguyen, H.T. Abba is an essential TRIM/RBCC protein to maintain the integrity of sarcomeric cytoarchitecture. J. Cell. Sci. 2013, 126, 3314–3323. [Google Scholar] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kejariwal, A.; Campbell, M.J.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; Muruganujan, A.; Rabkin, S.; et al. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003, 31, 334–341. [Google Scholar] [CrossRef]
- R_core_Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pages, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, C.; Rube, H.T.; Kribelbauer, J.F.; Crocker, J.; Loker, R.E.; Martini, G.D.; Laptenko, O.; Freed-Pastor, W.A.; Prives, C.; Stern, D.L.; et al. Accurate and sensitive quantification of protein-DNA binding affinity. Proc. Natl. Acad. Sci. USA 2018, 115, E3692–E3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlyueva, D.; Meireles-Filho, A.C.; Pagani, M.; Stark, A. Genome-Wide Ultrabithorax Binding Analysis Reveals Highly Targeted Genomic Loci at Developmental Regulators and a Potential Connection to Polycomb-Mediated Regulation. PLoS ONE 2016, 11, e0161997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erceg, J.; Pakozdi, T.; Marco-Ferreres, R.; Ghavi-Helm, Y.; Girardot, C.; Bracken, A.P.; Furlong, E.E. Dual functionality of cis-regulatory elements as developmental enhancers and Polycomb response elements. Genes Dev. 2017, 31, 590–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domsch, K.; Schroder, J.; Janeschik, M.; Schaub, C.; Lohmann, I. The Hox Transcription Factor Ubx Ensures Somatic Myogenesis by Suppressing the Mesodermal Master Regulator Twist. Cell Rep. 2021, 34, 108577. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folkendt, L.; Lohmann, I.; Domsch, K. An Evolutionary Perspective on Hox Binding Site Preferences in Two Different Tissues. J. Dev. Biol. 2021, 9, 57. https://doi.org/10.3390/jdb9040057
Folkendt L, Lohmann I, Domsch K. An Evolutionary Perspective on Hox Binding Site Preferences in Two Different Tissues. Journal of Developmental Biology. 2021; 9(4):57. https://doi.org/10.3390/jdb9040057
Chicago/Turabian StyleFolkendt, Laura, Ingrid Lohmann, and Katrin Domsch. 2021. "An Evolutionary Perspective on Hox Binding Site Preferences in Two Different Tissues" Journal of Developmental Biology 9, no. 4: 57. https://doi.org/10.3390/jdb9040057
APA StyleFolkendt, L., Lohmann, I., & Domsch, K. (2021). An Evolutionary Perspective on Hox Binding Site Preferences in Two Different Tissues. Journal of Developmental Biology, 9(4), 57. https://doi.org/10.3390/jdb9040057