
The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development


Abstract
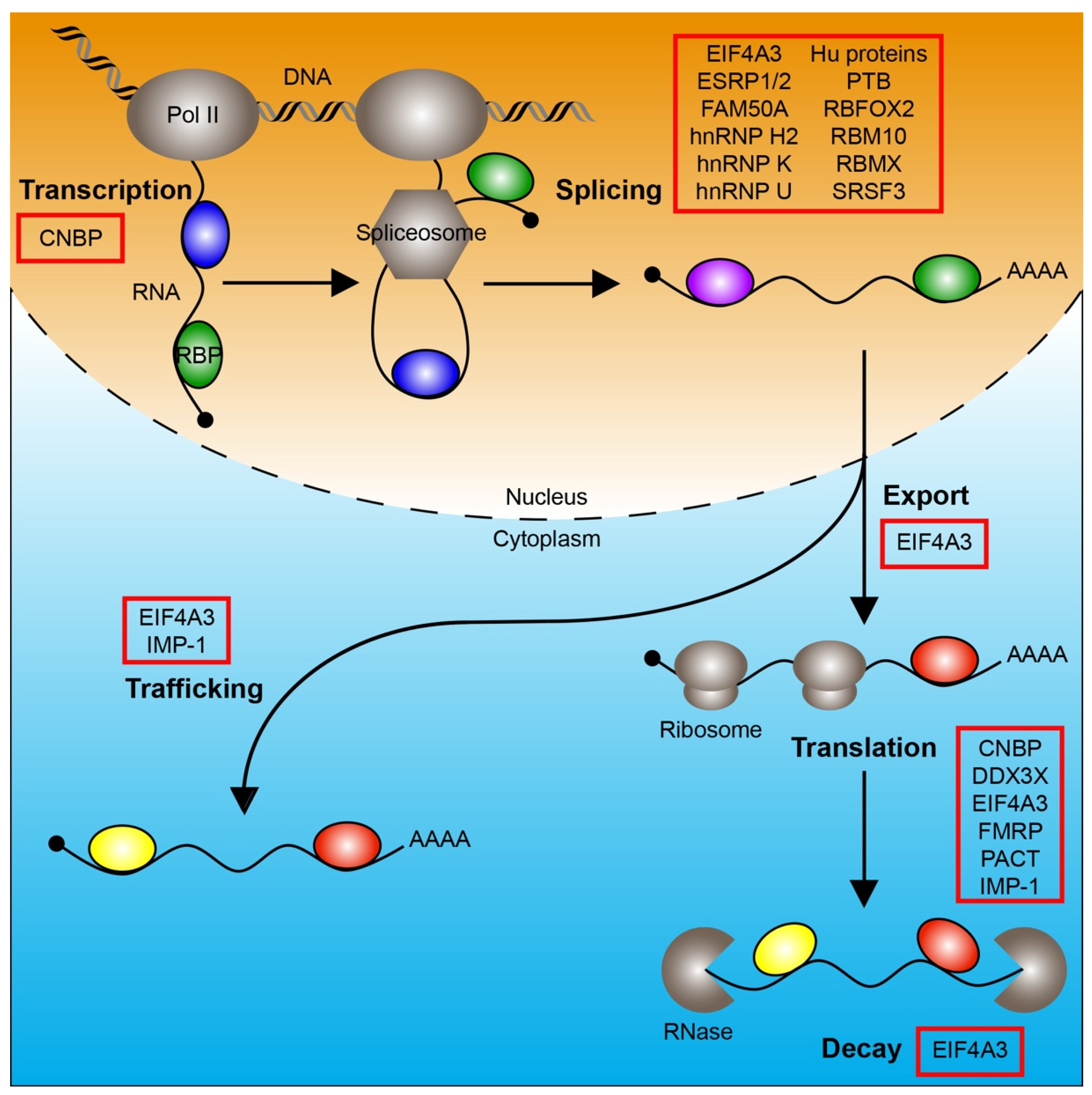
1. Introduction
2. RBPs Involved in Transcription
CNBP
3. RBPs Involved in Splicing
3.1. EIF4A3
3.2. ESRP1/2
3.3. FAM50A
3.4. hnRNP H2
3.5. hnRNP K
3.6. hnRNP U
3.7. Hu Proteins
3.8. PTB
3.9. RBFOX2
3.10. RBM10
3.11. RBMX
3.12. SRSF3
4. RBPs Involved in Trafficking
VICKZ Proteins
5. RBPs Involved in Translation
5.1. DDX3X
5.2. FMRP
5.3. PACT
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mayor, R.; Theveneau, E. The neural crest. Development 2013, 140, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2020, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Beauchamp, M.; Alam, S.S.; Kumar, S.; Jerome-Majewska, L.A. Spliceosomopathies and neurocristopathies: Two sides of the same coin? Dev. Dyn. 2020, 249, 924–945. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Thornton, C.A.; Griggs, R.C.; Moxley, R.T. Myotonic dystrophy with no trinucleotide repeat expansion. Ann. Neurol. 1994, 35, 269–272. [Google Scholar] [CrossRef]
- Udd, B.; Meola, G.; Krahe, R.; Thornton, C.; Ranum, L.; Bassez, G.; Kress, W.; Schoser, B.; Moxley, R. 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul. Disord. 2006, 16, 403–413. [Google Scholar] [CrossRef]
- Chen, W.; Liang, Y.; Deng, W.; Shimizu, K.; Ashique, A.M.; Li, E.; Li, Y.-P. The zinc-finger protein CNBP is required for forebrain formation in the mouse. Development 2003, 130, 1367–1379. [Google Scholar] [CrossRef]
- Shimizu, K.; Chen, W.; Ashique, A.M.; Moroi, R.; Li, Y.-P. Molecular cloning, developmental expression, promoter analysis and functional characterization of the mouse CNBP gene. Gene 2003, 307, 51–62. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Li, Y.-P. Haploinsuffciency for Znf9 in Znf9+/− Mice Is Associated with Multiorgan Abnormalities Resembling Myotonic Dystrophy. J. Mol. Biol. 2007, 368, 8–17. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of Cellular Nucleic Acid Binding Protein Encoded by a Myotonic Dystrophy Type 2 Gene Causes Muscle Atrophy. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Abe, Y.; Chen, W.; Huang, W.; Nishino, M.; Li, Y.-P. CNBP regulates forebrain formation at organogenesis stage in chick embryos. Dev. Biol. 2006, 295, 116–127. [Google Scholar] [CrossRef][Green Version]
- Flink, I.L.; Blitz, I.; Morkin, E. Characterization of cellular nucleic acid binding protein from Xenopus laevis: Expression in all three germ layers during early development. Dev. Dyn. 1998, 211, 123–130. [Google Scholar] [CrossRef]
- Armas, P.; Aguero, T.H.; Borgognone, M.; Aybar, M.J.; Calcaterra, N.B. Dissecting CNBP, a Zinc-Finger Protein Required for Neural Crest Development, in Its Structural and Functional Domains. J. Mol. Biol. 2008, 382, 1043–1056. [Google Scholar] [CrossRef]
- Weiner, A.; Allende, M.; Becker, T.; Calcaterra, N.B. CNBP mediates neural crest cell expansion by controlling cell proliferation and cell survival during rostral head development. J. Cell. Biochem. 2007, 102, 1553–1570. [Google Scholar] [CrossRef]
- Weiner, A.M.; Allende, M.L.; Calcaterra, N.B. Zebrafish cnbp intron1 plays a fundamental role in controlling spatiotemporal gene expression during embryonic development. J. Cell. Biochem. 2009, 108, 1364–1375. [Google Scholar] [CrossRef]
- Weiner, A.M.J.; Sdrigotti, M.A.; Kelsh, R.N.; Calcaterra, N.B. Deciphering the cellular and molecular roles of cellular nucleic acid binding protein during cranial neural crest development. Dev. Growth Differ. 2011, 53, 934–947. [Google Scholar] [CrossRef]
- De Peralta, M.S.P.; Mouguelar, V.S.; Sdrigotti, M.A.; Ishiy, F.A.A.; Fanganiello, R.D.; Passos-Bueno, M.R.; Coux, G.; Calcaterra, N.B. Cnbp ameliorates Treacher Collins Syndrome craniofacial anomalies through a pathway that involves redox-responsive genes. Cell Death Dis. 2016, 7, e2397. [Google Scholar] [CrossRef] [PubMed]
- Favaro, F.P.; Alvizi, L.; Zechi-Ceide, R.M.; Bertola, D.; Felix, T.M.; de Souza, J.; Raskin, S.; Twigg, S.; Weiner, A.M.; Armas, P.; et al. A Noncoding Expansion in EIF4A3 Causes Richieri-Costa-Pereira Syndrome, a Craniofacial Disorder Associated with Limb Defects. Am. J. Hum. Genet. 2014, 94, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Richieri-Costa, A.; Pereira, S.C.S. Short stature, Robin sequence, cleft mandible, pre/postaxial hand anomalies, and clubfoot: A new autosomal recessive syndrome. Am. J. Med. Genet. 1992, 42, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Favaro, F.P.; Zechi-Ceide, R.M.; Alvarez, C.W.; Maximino, L.; Antunes, L.F.B.B.; Richieri-Costa, A.; Guion-Almeida, M.L. Richieri-Costa-Pereira syndrome: A unique acrofacial dysostosis type. An overview of the Brazilian cases. Am. J. Med. Genet. Part A 2010, 155, 322–331. [Google Scholar] [CrossRef]
- Miller, E.E.; Kobayashi, G.; Musso, C.M.; Allen, M.; Ishiy, F.A.; De Caires, L.C.; Goulart, E.; Griesi-Oliveira, K.; Zechi-Ceide, R.M.; Richieri-Costa, A.; et al. EIF4A3 deficient human iPSCs and mouse models demonstrate neural crest defects that underlie Richieri-Costa-Pereira syndrome. Hum. Mol. Genet. 2017, 26, 2177–2191. [Google Scholar] [CrossRef] [PubMed]
- Rohacek, A.; Bebee, T.W.; Tilton, R.K.; Radens, C.; McDermott-Roe, C.; Peart, N.; Kaur, M.; Zaykaner, M.; Cieply, B.; Musunuru, K.; et al. ESRP1 Mutations Cause Hearing Loss due to Defects in Alternative Splicing that Disrupt Cochlear Development. Dev. Cell 2017, 43, 318–331.e5. [Google Scholar] [CrossRef]
- Cox, L.L.; Cox, T.C.; Uribe, L.M.M.; Zhu, Y.; Richter, C.T.; Nidey, N.; Standley, J.M.; Deng, M.; Blue, E.; Chong, J.; et al. Mutations in the Epithelial Cadherin-p120-Catenin Complex Cause Mendelian Non-Syndromic Cleft Lip with or without Cleft Palate. Am. J. Hum. Genet. 2018, 102, 1143–1157. [Google Scholar] [CrossRef]
- Revil, T.; Jerome-Majewska, L.A. During Embryogenesis, Esrp1 Expression Is Restricted to a Subset of Epithelial Cells and Is Associated With Splicing of a Number of Developmentally Important Genes. Dev. Dyn. 2013, 242, 281–290. [Google Scholar] [CrossRef]
- Carroll, S.H.; Trevino, C.M.; Li, E.B.; Kawasaki, K.; Myers, N.; Hallett, S.A.; Alhazmi, N.; Cotney, J.; Carstens, R.P.; Liao, E.C. An Irf6-Esrp1/2 regulatory axis controls midface morphogenesis in vertebrates. Development 2020, 147. [Google Scholar] [CrossRef]
- Warzecha, C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 Are Epithelial Cell-Type-Specific Regulators of FGFR2 Splicing. Mol. Cell 2009, 33, 591–601. [Google Scholar] [CrossRef]
- Bebee, T.W.; Park, J.W.; Sheridan, K.I.; Warzecha, C.; Cieply, B.W.; Rohacek, A.; Xing, Y.; Carstens, R.P. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 2015, 4, e08954. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sears, M.J.; Zhang, Z.; Li, H.; Salhab, I.; Krebs, P.; Xing, Y.; Nah, H.-D.; Williams, T.; Carstens, R.P. Cleft lip and cleft palate (CL/P) in Esrp1 KO mice is associated with alterations in epithelial-mesenchymal crosstalk. Development 2020, 147. [Google Scholar] [CrossRef]
- Burguera, D.; Marquez, Y.; Racioppi, C.; Permanyer, J.; Torres-Méndez, A.; Esposito, R.; Albuixech-Crespo, B.; Fanlo, L.; D’Agostino, Y.; Gohr, A.; et al. Evolutionary recruitment of flexible Esrp-dependent splicing programs into diverse embryonic morphogenetic processes. Nat. Commun. 2017, 8, 1799. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Khan, K.; Armfield-Uhas, K.; Srikanth, S.; Thompson, N.A.; Pardo, M.; Yu, L.; Norris, J.W.; Peng, Y.; Gripp, K.W.; et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat. Commun. 2020, 11, 3698. [Google Scholar] [CrossRef]
- Armfield, K.; Nelson, R.; Lubs, H.A.; Häne, B.; Schroer, R.J.; Arena, F.; Schwartz, C.E.; Stevenson, R.E. X-linked mental retardation syndrome with short stature, small hands and feet, seizures, cleft palate, and glaucoma is linked to Xq28. Am. J. Med. Genet. 1999, 85, 236–242. [Google Scholar] [CrossRef]
- Bain, J.M.; Cho, M.T.; Telegrafi, A.; Wilson, A.; Brooks, S.; Botti, C.; Gowans, G.; Autullo, L.A.; Krishnamurthy, V.; Willing, M.C.; et al. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am. J. Hum. Genet. 2016, 99, 728–734. [Google Scholar] [CrossRef]
- Bain, J.M.; Thornburg, O.; Pan, C.; Rome-Martin, D.; Boyle, L.; Fan, X.; Devinsky, O.; Frye, R.; Hamp, S.; Keator, C.G.; et al. Detailed Clinical and Psychological Phenotype of the X-linked HNRNPH2-Related Neurodevelopmental Disorder. Neurol. Genet. 2021, 7, e551. [Google Scholar] [CrossRef] [PubMed]
- Au, P.Y.B.; You, J.; Caluseriu, O.; Schwartzentruber, J.; Majewski, J.; Bernier, F.P.; Marcia Marcia Ferguson Care for Rare Canada Consortium; Valle, D.; Parboosingh, J.S.; Sobreira, N.; et al. GeneMatcher Aids in the Identification of a New Malformation Syndrome with Intellectual Disability, Unique Facial Dysmorphisms, and Skeletal and Connective Tissue Abnormalities Caused by De Novo Variants in HNRNPK. Hum. Mutat. 2015, 36, 1009–1014. [Google Scholar] [CrossRef]
- Au, P.Y.B.; Care for Rare Canada Consortium; Goedhart, C.; Ferguson, M.; Breckpot, J.; Devriendt, K.; Wierenga, K.; Fanning, E.; Grange, R.K.; Graham, G.E.; et al. Phenotypic spectrum of Au-Kline syndrome: A report of six new cases and review of the literature. Eur. J. Hum. Genet. 2018, 26, 1272–1281. [Google Scholar] [CrossRef]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Inaba, M.; Mizuno, S.; Shiina, M.; Imagawa, E.; Miyatake, S.; Nakashima, M.; Mizuguchi, T.; Takata, A.; Ogata, K.; et al. A case of atypical Kabuki syndrome arising from a novel missense variant in HNRNPK. Clin. Genet. 2017, 92, 554–555. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.; O’Roak, B.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De Novo Mutations in Moderate or Severe Intellectual Disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef]
- De Kovel, C.G.; Brilstra, E.H.; Van Kempen, M.J.; Slot, R.V.; Nijman, I.J.; Afawi, Z.; De Jonghe, P.; Djémié, T.; Guerrini, R.; Hardies, K.; et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol. Genet. Genom. Med. 2016, 4, 568–580. [Google Scholar] [CrossRef]
- Katsanou, V.; Milatos, S.; Yiakouvaki, A.; Sgantzis, N.; Kotsoni, A.; Alexiou, M.; Harokopos, V.; Aidinis, V.; Hemberger, M.; Kontoyiannis, D.L. The RNA-Binding Protein Elavl1/HuR Is Essential for Placental Branching Morphogenesis and Embryonic Development. Mol. Cell. Biol. 2009, 29, 2762–2776. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, W.; Fujihara, H.; Mitsuhashi, T.; Yano, M.; Shibata, S.; Hayakawa, Y.; Okano, H.J.; Sakakibara, S.-I.; Takano, H.; Takano, T.; et al. The RNA-binding protein HuD regulates neuronal cell identity and maturation. Proc. Natl. Acad. Sci. USA 2005, 102, 4625–4630. [Google Scholar] [CrossRef]
- Marusich, M.F.; Furneaux, H.M.; Henion, P.D.; Weston, J.A. Hu neuronal proteins are expressed in proliferating neurogenic cells. J. Neurobiol. 1994, 25, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, T.; Tokunaga, A.; Sakamoto, R.; Sagara, H.; Noguchi, S.; Sasaoka, T.; Yoshida, N. PTB Deficiency Causes the Loss of Adherens Junctions in the Dorsal Telencephalon and Leads to Lethal Hydrocephalus. Cereb. Cortex 2012, 23, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Noiret, M.; Audic, Y.; Hardy, S. Expression analysis of the polypyrimidine tract binding protein (PTBP1) and its paralogs PTBP2 and PTBP3 during Xenopus tropicalis embryogenesis. Int. J. Dev. Biol. 2012, 56, 747–753. [Google Scholar] [CrossRef]
- Cibi, D.M.; Mia, M.M.; Shekeran, S.G.; Yun, L.S.; Sandireddy, R.; Gupta, P.; Hota, M.; Sun, L.; Ghosh, S.; Singh, M.K. Neural crest-specific deletion of Rbfox2 in mice leads to craniofacial abnormalities including cleft palate. eLife 2019, 8, e45418. [Google Scholar] [CrossRef]
- Johnston, J.J.; Teer, J.K.; Cherukuri, P.F.; Hansen, N.F.; Loftus, S.; Chong, K.; Mullikin, J.C.; Biesecker, L.G. Massively Parallel Sequencing of Exons on the X Chromosome Identifies RBM10 as the Gene that Causes a Syndromic Form of Cleft Palate. Am. J. Hum. Genet. 2010, 86, 743–748. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Cervenka, J.; Anderson, R.C.; Sauk, J.J.; Bevis, W.D. Robin’s syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am. J. Dis. Child. 1970, 119, 176–178. [Google Scholar] [CrossRef]
- Kaeppler, K.E.; Stetson, R.C.; Lanpher, B.C.; Collura, C.A. Infant male with TARP syndrome: Review of clinical features, prognosis, and commonalities with previously reported patients. Am. J. Med. Genet. Part A 2018, 176, 2911–2914. [Google Scholar] [CrossRef]
- Kumps, C.; D’Haenens, E.; Vergult, S.; Leus, J.; van Coster, R.; Jansen, A.; Devriendt, K.; Oostra, A.; Vanakker, O.M. Phenotypic spectrum of the RBM10 -mediated intellectual disability and congenital malformation syndrome beyond classic TARP syndrome features. Clin. Genet. 2020, 99, 449–456. [Google Scholar] [CrossRef]
- Rodor, J.; FitzPatrick, D.R.; Eyras, E.; Cáceres, J.F. The RNA-binding landscape of RBM10 and its role in alternative splicing regulation in models of mouse early development. RNA Biol. 2016, 14, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; Xie, P.; Schoch, K.; Goldstein, D.B.; Howard, T.D.; Berry, M.N.; Schwartz, C.E.; Cronin, K.; Sliwa, S.; Allen, A.; et al. The RBMX gene as a candidate for the Shashi X-linked intellectual disability syndrome. Clin. Genet. 2014, 88, 386–390. [Google Scholar] [CrossRef]
- Shashi, V.; Berry, M.N.; Shoaf, S.; Sciote, J.J.; Goldstein, D.; Hart, T.C. A Unique Form of Mental Retardation with a Distinctive Phenotype Maps to Xq26-q27. Am. J. Hum. Genet. 2000, 66, 469–479. [Google Scholar] [CrossRef][Green Version]
- Dichmann, D.S.; Fletcher, R.B.; Harland, R.M. Expression cloning in Xenopus identifies RNA-binding proteins as regulators of embryogenesis and Rbmx as necessary for neural and muscle development. Dev. Dyn. 2008, 237, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Tsend-Ayush, E.; O’Sullivan, L.A.; Grutzner, F.; Onnebo, S.M.; Lewis, R.S.; Delbridge, M.L.; Graves, J.; Ward, A. RBMX gene is essential for brain development in zebrafish. Dev. Dyn. 2005, 234, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Dennison, B.J.C.; Larson, E.D.; Fu, R.; Mo, J.; Fantauzzo, K.A. Srsf3 mediates alternative RNA splicing downstream of PDGFRα signaling in the facial mesenchyme. Development 2021, 148. [Google Scholar] [CrossRef] [PubMed]
- Klinghoffer, R.A.; Hamilton, T.; Hoch, R.; Soriano, P. An Allelic Series at the PDGFαR Locus Indicates Unequal Contributions of Distinct Signaling Pathways During Development. Dev. Cell 2002, 2, 103–113. [Google Scholar] [CrossRef]
- Fantauzzo, K.A.; Soriano, P. PI3K-mediated PDGFRα signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 2014, 28, 1005–1017. [Google Scholar] [CrossRef]
- Hansen, T.V.O.; Hammer, N.A.; Nielsen, J.; Madsen, M.; Dalbaeck, C.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Dwarfism and Impaired Gut Development in Insulin-Like Growth Factor II mRNA-Binding Protein 1-Deficient Mice. Mol. Cell. Biol. 2004, 24, 4448–4464. [Google Scholar] [CrossRef] [PubMed]
- Carmel, M.S.; Kahane, N.; Oberman, F.; Miloslavski, R.; Sela-Donenfeld, D.; Kalcheim, C.; Yisraeli, J.K. A Novel Role for VICKZ Proteins in Maintaining Epithelial Integrity during Embryogenesis. PLoS ONE 2015, 10, e0136408. [Google Scholar] [CrossRef]
- Blok, L.S.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef]
- Chanes, N.M.; Wong, J.; Lacassie, Y. Further delineation of DDX3X syndrome. Clin. Dysmorphol. 2019, 28, 149–151. [Google Scholar] [CrossRef]
- Lennox, A.L.; Hoye, M.L.; Jiang, R.; Johnson-Kerner, B.L.; Suit, L.A.; Venkataramanan, S.; Sheehan, C.; Alsina, F.C.; Fregeau, B.; Aldinger, K.; et al. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron 2020, 106, 404–420.e8. [Google Scholar] [CrossRef] [PubMed]
- Beal, B.; Hayes, I.; McGaughran, J.; Amor, D.J.; Miteff, C.; Jackson, V.; Van Reyk, O.; Subramanian, G.; Hildebrand, M.S.; Morgan, A.; et al. Expansion of phenotype of DDX3X syndrome: Six new cases. Clin. Dysmorphol. 2019, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Kellaris, G.; Khan, K.; Baig, S.M.; Tsai, I.-C.; Zamora, F.M.; Ruggieri, P.; Natowicz, M.R.; Katsanis, N. A hypomorphic inherited pathogenic variant in DDX3X causes male intellectual disability with additional neurodevelopmental and neurodegenerative features. Hum. Genom. 2018, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Nicola, P.; Blackburn, P.R.; Rasmussen, K.J.; Bertsch, N.L.; Klee, E.W.; Hasadsri, L.; Pichurin, P.N.; Rankin, J.; Raymond, F.L.; Clayton-Smith, J.; et al. De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability. Am. J. Med. Genet. Part A 2019, 179, 570–578. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chan, C.-H.; Chen, C.-M.; Tsai, Y.-S.; Tsai, T.-Y.; Lee, Y.-H.W.; You, L.-R. Targeted inactivation of murine Ddx3x: Essential roles of Ddx3x in placentation and embryogenesis. Hum. Mol. Genet. 2016, 25, 2905–2922. [Google Scholar] [CrossRef][Green Version]
- Perfetto, M.; Xu, X.; Lu, C.; Shi, Y.; Yousaf, N.; Li, J.; Yien, Y.Y.; Wei, S. The RNA helicase DDX3 induces neural crest by promoting AKT activity. Development 2020, 148, dev184341. [Google Scholar] [CrossRef]
- The Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature 2014, 519, 223–228. [CrossRef]
- Cruciat, C.-M.; Dolde, C.; de Groot, R.E.A.; Ohkawara, B.; Reinhard, C.; Korswagen, H.C.; Niehrs, C. RNA Helicase DDX3 Is a Regulatory Subunit of Casein Kinase 1 in Wnt-β-Catenin Signaling. Science 2013, 339, 1436–1441. [Google Scholar] [CrossRef]
- Kremer, E.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Devys, D.; Biancalana, V.; Rousseau, F.; Boué, J.; Mandel, J.L.; Oberlé, I. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am. J. Med. Genet. 1992, 43, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Blanchet, P.; Kobetz, A.; Marchant, D.; Faucon, N.; Sarda, P.; Moraine, C.; Sittler, A.; Biancalana, V.; Malafosse, A.; et al. Expression of FMR1, FXR1, and FXR2 genes in human prenatal tissues. J. Neuropathol. Exp. Neurol. 1999, 58, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.P.; Bell, J. A PEDIGREE OF MENTAL DEFECT SHOWING SEX-LINKAGE. J. Neurol. Neurosurg. Psychiatry 1943, 6, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Mattei, J.F.; Mattei, M.-G.; Aumeras, C.; Auger, M.; Giraud, F. X-linked mental retardation with the fragile X. A study of 15 families. Qual. Life Res. 1981, 59, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Allen, G.A.; Singh, D.N.; Carpenter, N.J.; Hall, B.D. Preliminary communication: Photoanthropometric analysis of individuals with the fragile X syndrome. Am. J. Med. Genet. 1988, 30, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Grønskov, K.; Brøndum-Nielsen, K.; Dedic, A.; Hjalgrim, H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur. J. Hum. Genet. 2011, 19, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Heulens, I.; Suttie, M.; Postnov, A.; De Clerck, N.; Perrotta, C.S.; Mattina, T.; Faravelli, F.; Forzano, F.; Kooy, F.; Hammond, P. Craniofacial characteristics of fragile X syndrome in mouse and man. Eur. J. Hum. Genet. 2012, 21, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Hinds, H.L.; Ashley, C.T.; Sutcliffe, J.S.; Nelson, D.L.; Warren, S.T.; Housman, D.E.; Schalling, M. Tissue specific expression of FMR–1 provides evidence for a functional role in fragile X syndrome. Nat. Genet. 1993, 3, 36–43. [Google Scholar] [CrossRef]
- Bakker, C.E.; Verheij, C.; Willemsen, R.; van der Helm, R.; Oerlemans, F.; Vermey, M.; Bygrave, A.; Hoogeveen, A.; Oostra, B.A.; Reyniers, E.; et al. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78, 23–33. [Google Scholar]
- Lim, J.H.; Booker, A.B.; Luo, T.; Williams, T.; Furuta, Y.; Lagutin, O.; Oliver, G.; Sargent, T.D.; Fallon, J.R. AP-2α selectively regulates fragile X mental retardation-1 gene transcription during embryonic development. Hum. Mol. Genet. 2005, 14, 2027–2034. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, J.H.; Luo, T.; Sargent, T.D.; Fallon, J.R. Developmental expression of Xenopus Fragile X mental retardation-1 gene. Int. J. Dev. Biol. 2005, 49, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Gessert, S.; Bugner, V.; Tecza, A.; Pinker, M.; Kühl, M. FMR1/FXR1 and the miRNA pathway are required for eye and neural crest development. Dev. Biol. 2010, 341, 222–235. [Google Scholar] [CrossRef]
- Tucker, B.; Richards, R.I.; Lardelli, M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 2006, 15, 3446–3458. [Google Scholar] [CrossRef]
- Broeder, M.J.D.; Van Der Linde, H.; Brouwer, J.R.; Oostra, B.A.; Willemsen, R.; Ketting, R.F. Generation and Characterization of Fmr1 Knockout Zebrafish. PLoS ONE 2009, 4, e7910. [Google Scholar] [CrossRef]
- Camargos, S.; Scholz, S.; Sánchez, J.S.; Paisán-Ruiz, C.; Lewis, P.; Hernandez, D.; Ding, J.; Gibbs, J.R.; Cookson, M.R.; Bras, J.; et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008, 7, 207–215. [Google Scholar] [CrossRef]
- Zech, M.; Castrop, F.; Schormair, B.; Jochim, A.; Wieland, T.; Gross, N.; Lichtner, P.; Peters, A.; Gieger, C.; Meitinger, T.; et al. DYT16 revisited: Exome sequencing identifies PRKRA mutations in a European dystonia family. Mov. Disord. 2014, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Rowe, T.M.; Rizzi, M.; Hirose, K.; Peters, G.; Sen, G.C. A role of the double-stranded RNA-binding protein PACT in mouse ear development and hearing. Proc. Natl. Acad. Sci. USA 2006, 103, 5823–5828. [Google Scholar] [CrossRef]
- Bennett, R.; Blalock, W.L.; Choi, E.-J.; Lee, Y.J.; Zhang, Y.; Zhou, L.; Oh, S.P.; May, W.S. RAX is required for fly neuronal development and mouse embryogenesis. Mech. Dev. 2008, 125, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Seachrist, D.D.; Keri, R.A.; Sen, G.C. The double-stranded RNA-binding protein, PACT, is required for postnatal anterior pituitary proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 10696–10701. [Google Scholar] [CrossRef]
- Dickerman, B.K.; White, C.L.; Chevalier, C.; Nalesso, V.; Charles, C.; Fouchécourt, S.; Guillou, F.; Viriot, L.; Sen, G.C.; Herault, Y. Missense Mutation in the Second RNA Binding Domain Reveals a Role for Prkra (PACT/RAX) during Skull Development. PLoS ONE 2011, 6, e28537. [Google Scholar] [CrossRef]
- Fowles, L.F.; Bennetts, J.S.; Berkman, J.L.; Williams, E.; Koopman, P.; Teasdale, R.; Wicking, C. Genomic screen for genes involved in mammalian craniofacial development. Genesis 2003, 35, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Jones, K.L.; Smith, F.J.; Williams, T.; Li, H. An Alternative Splicing Program for Mouse Craniofacial Development. Front. Physiol. 2020, 11, 1099. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; McMahon, J.J.; Tsai, Y.-H.; Wang, Z.; Silver, D.L. Haploinsufficiency for Core Exon Junction Complex Components Disrupts Embryonic Neurogenesis and Causes p53-Mediated Microcephaly. PLoS Genet. 2016, 12, e1006282. [Google Scholar] [CrossRef]


{kind=link}
| Disorder (Associated RBP) | Craniofacial Defect(s) |
|---|---|
| DM2 (CNBP) | Hearing loss |
| MRXSSB (DDX3X) | Females: microcephaly; dolichocephaly; long and/or hypotonic face; high and/or broad forehead; hypertelorism; visual impairment; wide nasal bridge; bulbous nose; narrow alae nasi; anteverted nostrils; cleft lip and/or palate; hearing loss Males: brachycephaly; macrocephaly; strabismus; bifid uvula; visual impairment; hearing loss |
| Robin sequence with cleft mandible and limb anomalies (EIF4A3) | Microstomia; cleft palate; absent lower central incisors; micrognathia; abnormal fusion of the mandible; ear anomalies |
| DFNB109 (ESRP1) | Hearing loss; vestibular dysplasia |
| Non-syndromic cleft lip with or without cleft palate (ESRP2) | Cleft lip and/or palate |
| MRXSA (FAM50A) | Macrocephaly; prominent, tall and/or broad forehead; bitemporal narrowing; faint hemangiomas between brows; downslanting palpebral fissures; hypotelorism; epicanthal folds; infraorbital creases; proptosis; exotropia; strabismus; keratoconus; nystagmus; depressed nasal bridge; wide nasal root; short and lightly upturned nose with underdeveloped nares; tubular nose; bulbous nose; prominent lips; bow-shaped mouth; cleft palate; hypodontia; microretrognathia; micrognathia; low-set, large and/or slightly posteriorly rotated ears; excessively folded helices |
| FXS (FMRP) | Long, narrow face; high and/or broad forehead; broad palpebral fissures; midface hypoplasia; flat nasal bridge; broad nose; upward displacement of the nasal alae; broad philtrum; large mouth; thick lips; long upper middle incisors; high-arched palate; prognathia; hypotonia affecting the lower jaw; large, low-set and/or poorly formed ears |
| MRXSB (hnRNP H2) | Microcephaly; short palpebral fissures; almond-shaped eyes; strabismus; long columella; hypoplastic alae nasi; short philtrum; full lower lip; micrognathia |
| AUKS (hnRNP K) | Dolichocephaly; craniosynostosis; ridged metopic suture; long face; broad or sparse eyebrows; long palpebral fissures; shallow orbits; ptosis; lateral eversion of the lower eyelids; vision abnormalities; full cheeks; broad nasal bridge; bifid nasal tip; hypoplastic alae nasi; open or down-turned mouth; thin upper lip; high or cleft palate; prominent midline groove of the tongue; missing teeth; malocclusion; low-set, prominent ears; underdeveloped ear helices; preauricular pits; earlobe crease; hearing loss |
| DEE54 (hnRNP U) | Microcephaly; deep-set eyes with epicanthal folds; gray sclerae; narrow palate |
| DYT16 (PACT) | Oromandibular dystonia; facial grimacing; blepharospasm; hypomimia |
| TARPS (RBM10) | Large fontanels; round face; hypertelorism; vision impairment; underdevelopment of the alae nasi; prominent columella; wide mouth; downturned mouth corners; high, narrow and/or cleft palate; glossoptosis; micrognathia; low-set and/or posteriorly rotated ears; hearing loss |
| MRXS11 (RBMX) | Coarse facies; prominent supraorbital ridges; narrow palpebral fissures; puffy eyelids; bulbous nose; prominent lower lip; large ears |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forman, T.E.; Dennison, B.J.C.; Fantauzzo, K.A. The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development. J. Dev. Biol. 2021, 9, 34. https://doi.org/10.3390/jdb9030034
Forman TE, Dennison BJC, Fantauzzo KA. The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development. Journal of Developmental Biology. 2021; 9(3):34. https://doi.org/10.3390/jdb9030034
Chicago/Turabian StyleForman, Thomas E., Brenna J. C. Dennison, and Katherine A. Fantauzzo. 2021. "The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development" Journal of Developmental Biology 9, no. 3: 34. https://doi.org/10.3390/jdb9030034
APA StyleForman, T. E., Dennison, B. J. C., & Fantauzzo, K. A. (2021). The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development. Journal of Developmental Biology, 9(3), 34. https://doi.org/10.3390/jdb9030034