Hedgehog Signaling and Embryonic Craniofacial Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
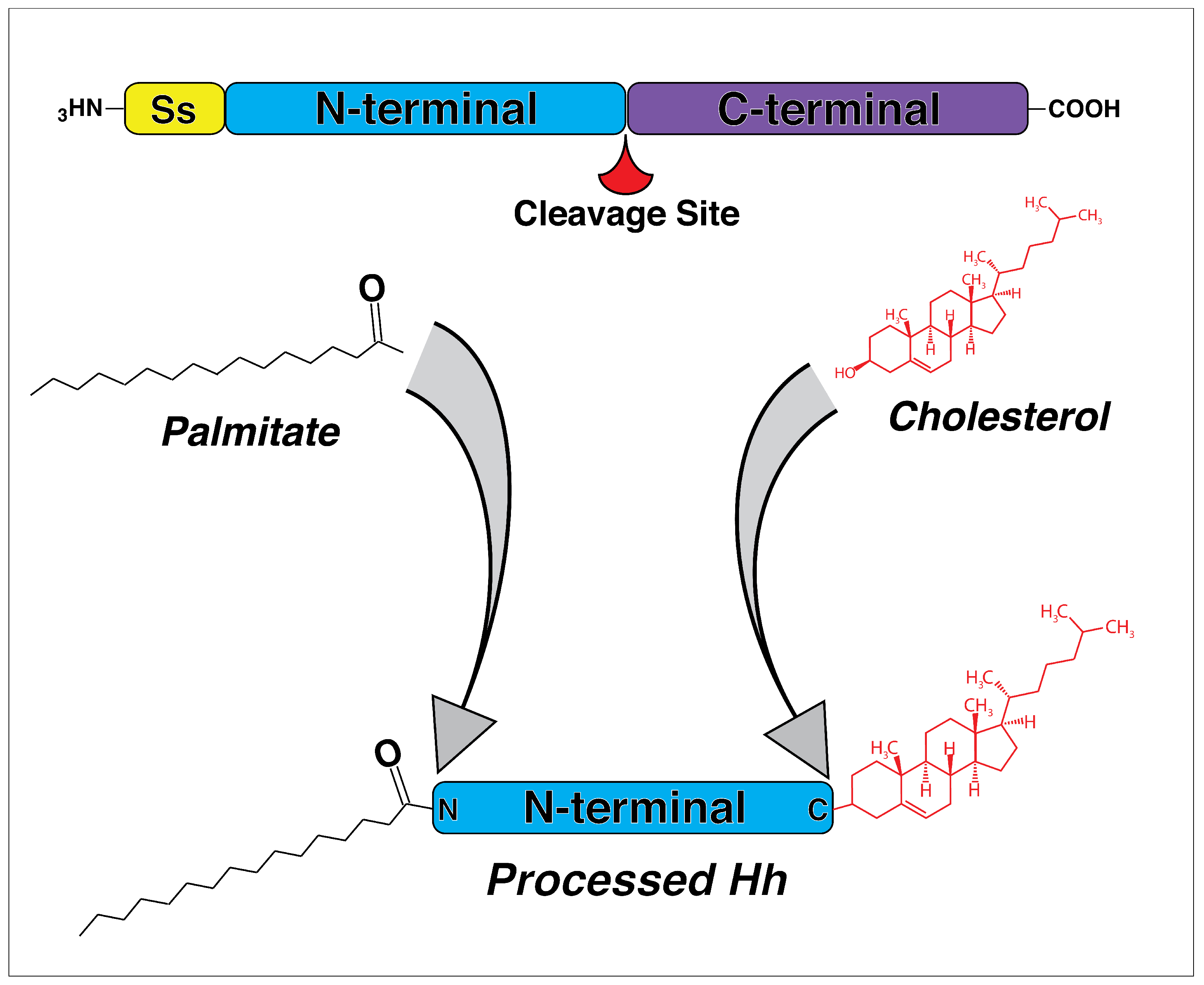
2. The Discovery and Evolution of Hedgehog Genes
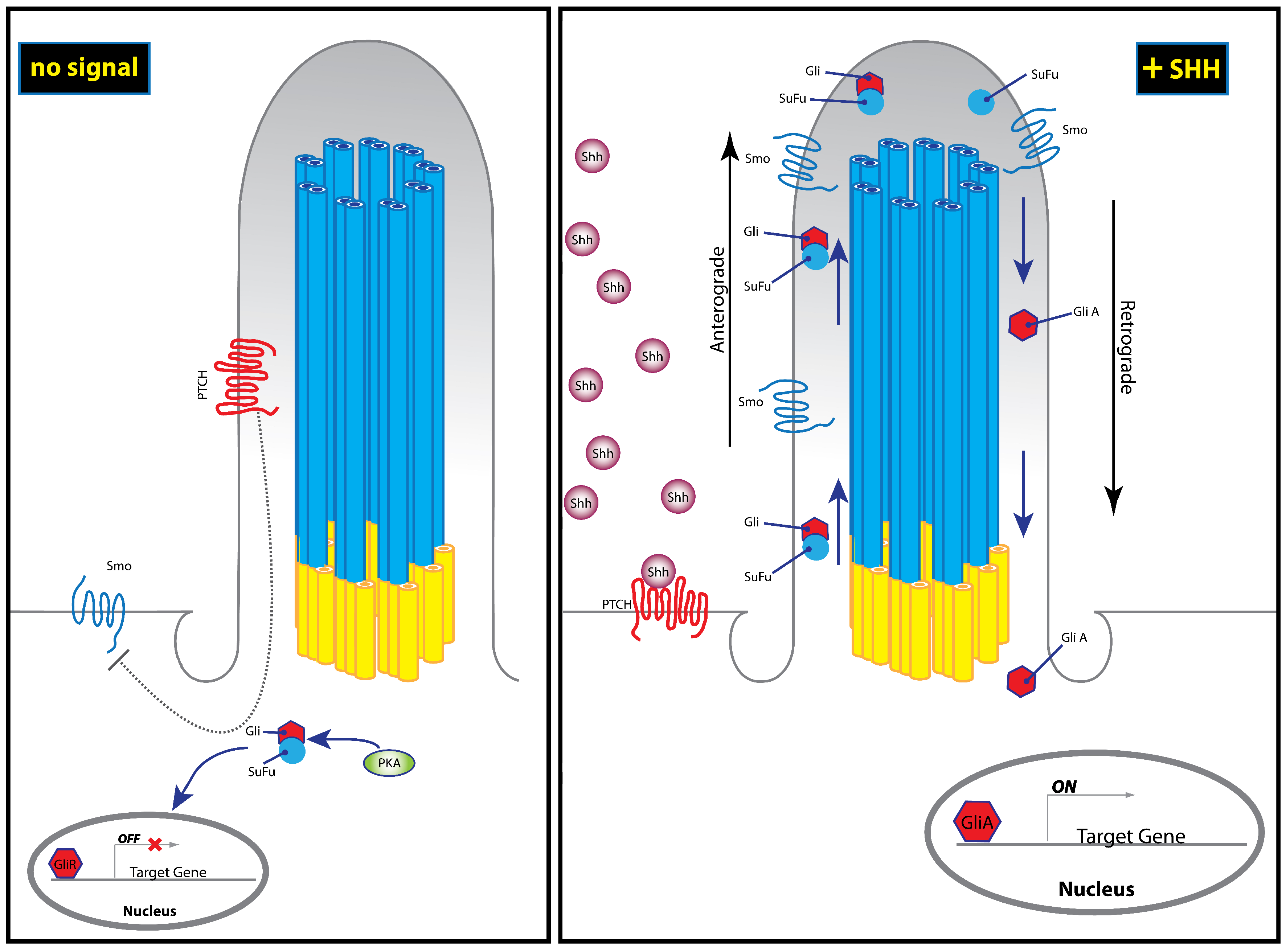
3. The Hedgehog Pathway
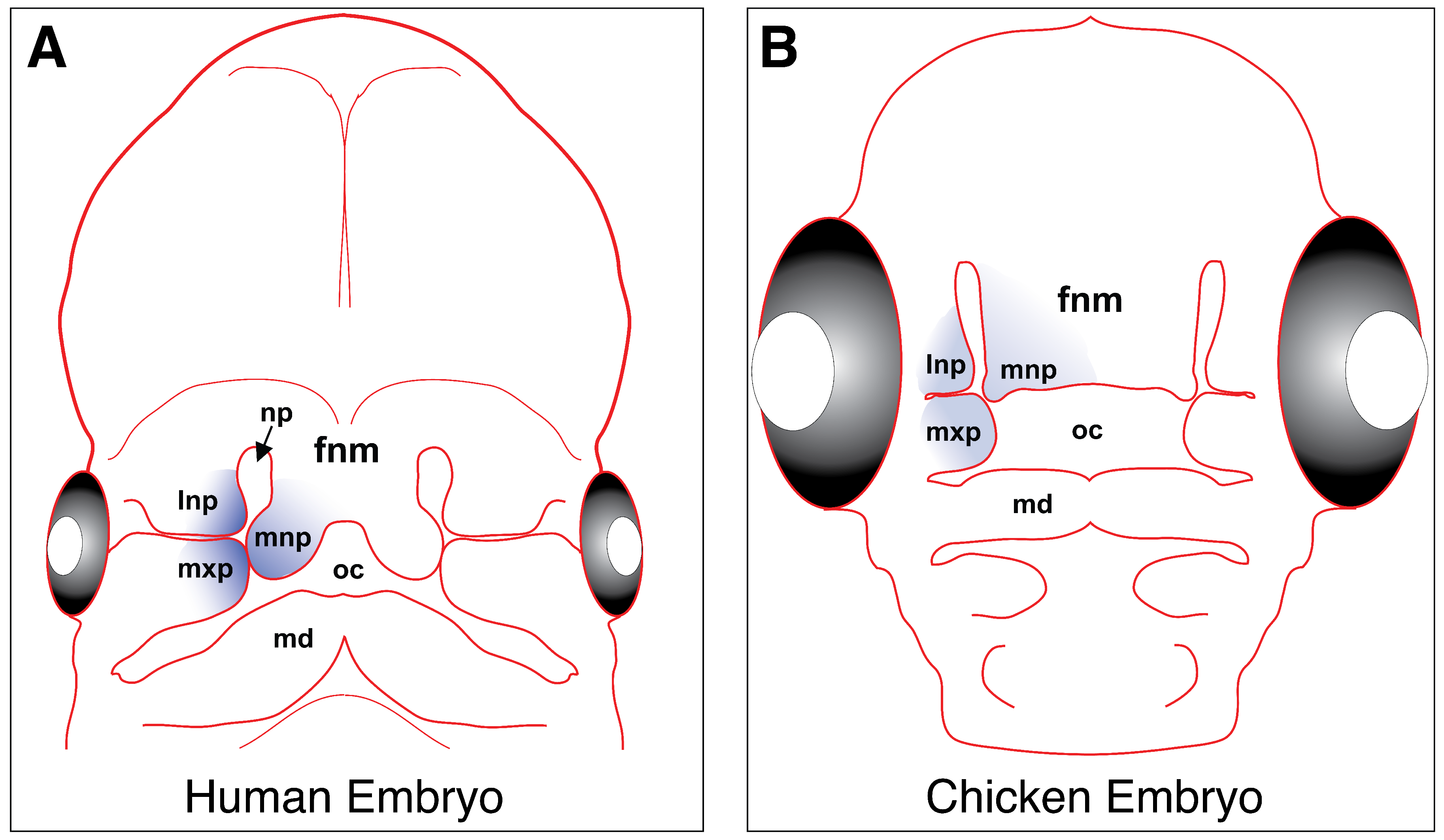
4. Craniofacial Development
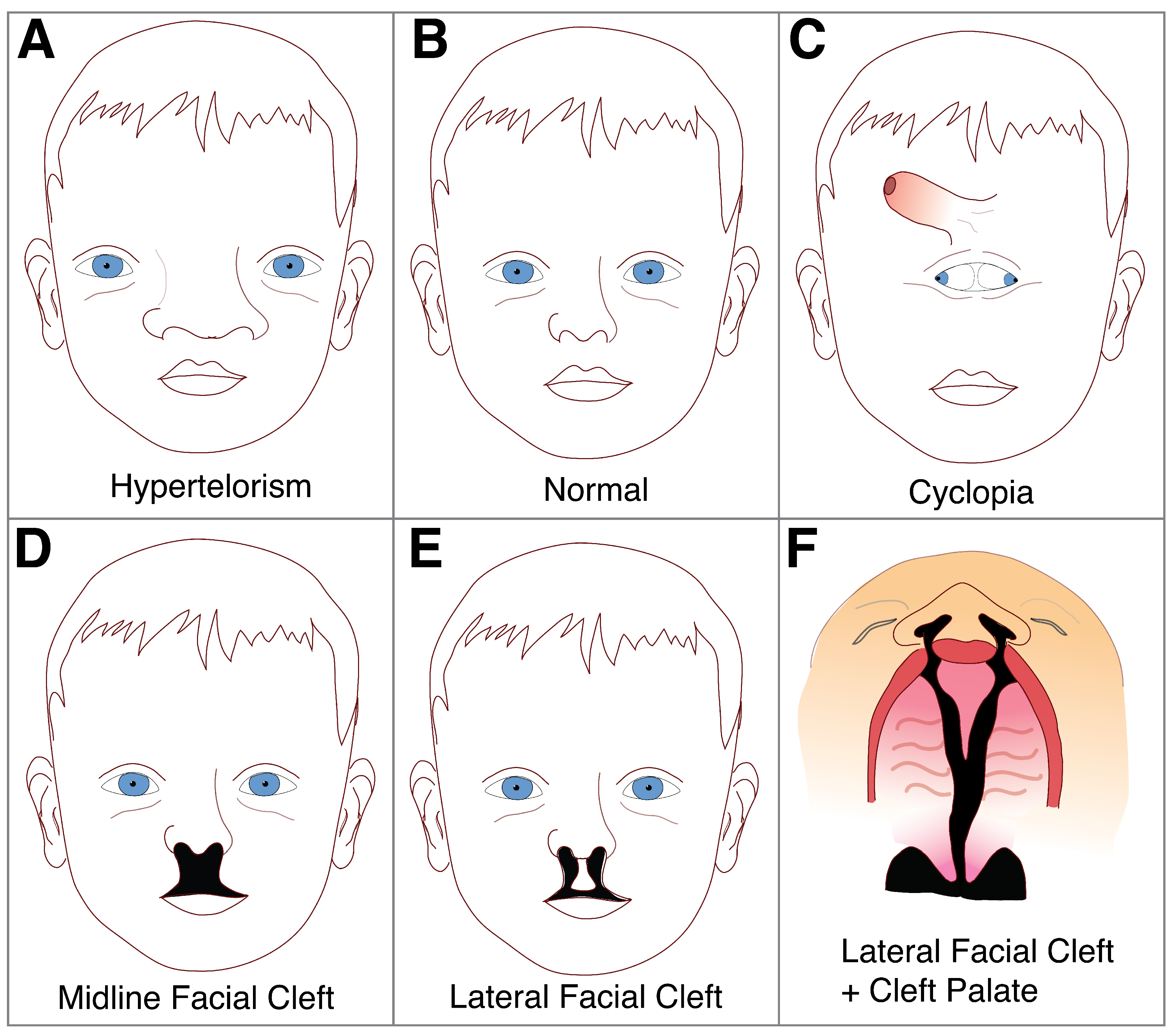
5. Disorders of the Midline: Hypertelorism and Holoprosencephaly
6. Cleft Lip and Palate
7. Ciliopathies
8. Talpid Chicken Mutants
9. Fetal Alcohol Syndrome
10. Statins and Cholesterol Biosynthesis
11. Craniosynostosis
12. Conclusions
Funding
Conflicts of Interest
References
- Schutte, B.C.; Murray, J.C. The many faces and factors of orofacial clefts. Hum. Mol. Genet. 1999, 8, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Ahlgren, S.C.; Bronner-Fraser, M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr. Biol. 1999, 9, 1304–1314. [Google Scholar] [CrossRef]
- Meyers, E.N.; Martin, G.R. Differences in left-right axis pathways in mouse and chick: Functions of FGF8 and SHH. Science 1999, 285, 403–406. [Google Scholar] [CrossRef]
- Schilling, T.F.; Concordet, J.P.; Ingham, P.W. Regulation of left-right asymmetries in the zebrafish by Shh and BMP4. Dev. Biol. 1999, 210, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Laufer, E.; Nelson, C.E.; Johnson, R.L.; Morgan, B.A.; Tabin, C. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell 1994, 79, 993–1003. [Google Scholar] [CrossRef]
- Johnson, R.L.; Laufer, E.; Riddle, R.D.; Tabin, C. Ectopic expression of Sonic hedgehog alters dorsal-ventral patterning of somites. Cell 1994, 79, 1165–1173. [Google Scholar] [CrossRef]
- Jensen, A.M.; Wallace, V.A. Expression of Sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development 1997, 124, 363–371. [Google Scholar] [PubMed]
- Levine, E.M.; Roelink, H.; Turner, J.; Reh, T.A. Sonic hedgehog promotes rod photoreceptor differentiation in mammalian retinal cells in vitro. J. Neurosci. 1997, 17, 6277–6288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Yang, X.J. Temporal and spatial effects of Sonic hedgehog signaling in chick eye morphogenesis. Dev. Biol. 2001, 233, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Teillet, M.; Watanabe, Y.; Jeffs, P.; Duprez, D.; Lapointe, F.; Le Douarin, N.M. Sonic hedgehog is required for survival of both myogenic and chondrogenic somitic lineages. Development 1998, 125, 2019–2030. [Google Scholar] [PubMed]
- Murtaugh, L.C.; Chyung, J.H.; Lassar, A.B. Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev. 1999, 13, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Munsterberg, A.E.; Kitajewski, J.; Bumcrot, D.A.; McMahon, A.P.; Lassar, A.B. Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev. 1995, 9, 2911–2922. [Google Scholar] [CrossRef]
- Blagden, C.S.; Currie, P.D.; Ingham, P.W.; Hughes, S.M. Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev. 1997, 11, 2163–2175. [Google Scholar] [CrossRef]
- Duprez, D.; Fournier-Thibault, C.; Le Douarin, N. Sonic Hedgehog induces proliferation of committed skeletal muscle cells in the chick limb. Development 1998, 125, 495–505. [Google Scholar]
- Borycki, A.G.; Brunk, B.; Tajbakhsh, S.; Buckingham, M.; Chiang, C.; Emerson, C.P., Jr. Sonic hedgehog controls epaxial muscle determination through Myf5 activation. Development 1999, 126, 4053–4063. [Google Scholar]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Roelink, H.; Augsburger, A.; Heemskerk, J.; Korzh, V.; Norlin, S.; Ruiz i Altaba, A.; Tanabe, Y.; Placzek, M.; Edlund, T.; Jessell, T.M.; et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 1994, 76, 761–775. [Google Scholar] [CrossRef]
- Roelink, H.; Porter, J.A.; Chiang, C.; Tanabe, Y.; Chang, D.T.; Beachy, P.A.; Jessell, T.M. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 1995, 81, 445–455. [Google Scholar] [CrossRef]
- Dassule, H.R.; McMahon, A.P. Analysis of epithelial-mesenchymal interactions in the initial morphogenesis of the mammalian tooth. Dev. Biol. 1998, 202, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, Z.; Hui, C.C.; Sharpe, P.T. The Shh signalling pathway in early tooth development. Cell. Mol. Biol. 1999, 45, 567–578. [Google Scholar] [PubMed]
- Pereira, J.; Johnson, W.E.; O’Brien, S.J.; Jarvis, E.D.; Zhang, G.; Gilbert, M.T.; Vasconcelos, V.; Antunes, A. Evolutionary genomics and adaptive evolution of the Hedgehog gene family (Shh, Ihh and Dhh) in vertebrates. PLoS ONE 2014, 9, e74132. [Google Scholar] [CrossRef] [PubMed]
- Cobourne, M.T.; Green, J.B. Hedgehog signalling in development of the secondary palate. Front. Oral Biol. 2012, 16, 52–59. [Google Scholar] [CrossRef]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog receptor function during craniofacial development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef]
- Dworkin, S.; Boglev, Y.; Owens, H.; Goldie, S.J. The Role of Sonic Hedgehog in Craniofacial Patterning, Morphogenesis and Cranial Neural Crest Survival. J. Dev. Biol. 2016, 4. [Google Scholar] [CrossRef]
- Kurosaka, H. The Roles of Hedgehog Signaling in Upper Lip Formation. Biomed Res. Int. 2015, 2015, 901041. [Google Scholar] [CrossRef]
- Seppala, M.; Fraser, G.J.; Birjandi, A.A.; Xavier, G.M.; Cobourne, M.T. Sonic Hedgehog Signaling and Development of the Dentition. J. Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Jiang, R. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 2009, 136, 1387–1396. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Yao, H.H.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef]
- Parmantier, E.; Lynn, B.; Lawson, D.; Turmaine, M.; Namini, S.S.; Chakrabarti, L.; McMahon, A.P.; Jessen, K.R.; Mirsky, R. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 1999, 23, 713–724. [Google Scholar] [CrossRef]
- Mirsky, R.; Parmantier, E.; McMahon, A.P.; Jessen, K.R. Schwann cell-derived desert hedgehog signals nerve sheath formation. Ann. N. Y. Acad. Sci. 1999, 883, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; von Kessler, D.P.; Parks, S.; Beachy, P.A. Secretion and localized transcription suggest a role in positional signaling for products of the segmentation gene hedgehog. Cell 1992, 71, 33–50. [Google Scholar] [CrossRef]
- Mohler, J.; Vani, K. Molecular organization and embryonic expression of the hedgehog gene involved in cell-cell communication in segmental patterning of Drosophila. Development 1992, 115, 957–971. [Google Scholar] [PubMed]
- Tabata, T.; Eaton, S.; Kornberg, T.B. The Drosophila hedgehog gene is expressed specifically in posterior compartment cells and is a target of engrailed regulation. Genes Dev. 1992, 6, 2635–2645. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Chang, D.T.; Lopez, A.; von Kessler, D.P.; Chiang, C.; Simandl, B.K.; Zhao, R.; Seldin, M.F.; Fallon, J.F.; Beachy, P.A. Products, genetic linkage and limb patterning activity of a murine hedgehog gene. Development 1994, 120, 3339–3353. [Google Scholar] [PubMed]
- Ekker, S.C.; Ungar, A.R.; Greenstein, P.; von Kessler, D.P.; Porter, J.A.; Moon, R.T.; Beachy, P.A. Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr. Biol. 1995, 5, 944–955. [Google Scholar] [CrossRef]
- Parkin, C.A.; Allen, C.E.; Ingham, P.W. Hedgehog signalling is required for cloacal development in the zebrafish embryo. Int. J. Dev. Biol. 2009, 53, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Currie, P.D.; Ingham, P.W. Induction of a specific muscle cell type by a hedgehog-like protein in zebrafish. Nature 1996, 382, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.; Nellen, D.; Bellotto, M.; Hafen, E.; Senti, K.A.; Dickson, B.J.; Basler, K. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 1999, 99, 803–815. [Google Scholar] [CrossRef]
- Ma, Y.; Erkner, A.; Gong, R.; Yao, S.; Taipale, J.; Basler, K.; Beachy, P.A. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 2002, 111, 63–75. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Kuzmickas, R.P.; Jao, C.Y.; Liu, J.; Salic, A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012, 2, 308–320. [Google Scholar] [CrossRef]
- Hall, T.M.; Porter, J.A.; Beachy, P.A.; Leahy, D.J. A potential catalytic site revealed by the 1.7-A crystal structure of the amino-terminal signalling domain of Sonic hedgehog. Nature 1995, 378, 212–216. [Google Scholar] [CrossRef]
- Porter, J.A.; Ekker, S.C.; Park, W.J.; von Kessler, D.P.; Young, K.E.; Chen, C.H.; Ma, Y.; Woods, A.S.; Cotter, R.J.; Koonin, E.V.; et al. Hedgehog patterning activity: Role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 1996, 86, 21–34. [Google Scholar] [CrossRef]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Vargas, F.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum. Mol. Genet. 1997, 6, 1847–1853. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Zeng, C.; Wen, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef]
- Rietveld, A.; Neutz, S.; Simons, K.; Eaton, S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J. Biol. Chem. 1999, 274, 12049–12054. [Google Scholar] [CrossRef]
- Chen, M.H.; Li, Y.J.; Kawakami, T.; Xu, S.M.; Chuang, P.T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004, 18, 641–659. [Google Scholar] [CrossRef]
- Zeng, X.; Goetz, J.A.; Suber, L.M.; Scott, W.J., Jr.; Schreiner, C.M.; Robbins, D.J. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 2001, 411, 716–720. [Google Scholar] [CrossRef]
- Guerrero, I.; Chiang, C. A conserved mechanism of Hedgehog gradient formation by lipid modifications. Trends Cell Biol. 2007, 17, 1–5. [Google Scholar] [CrossRef]
- Caspary, T.; Garcia-Garcia, M.J.; Huangfu, D.; Eggenschwiler, J.T.; Wyler, M.R.; Rakeman, A.S.; Alcorn, H.L.; Anderson, K.V. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr. Biol. 2002, 12, 1628–1632. [Google Scholar] [CrossRef]
- Creanga, A.; Glenn, T.D.; Mann, R.K.; Saunders, A.M.; Talbot, W.S.; Beachy, P.A. Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev. 2012, 26, 1312–1325. [Google Scholar] [CrossRef]
- Goodrich, L.V.; Johnson, R.L.; Milenkovic, L.; McMahon, J.A.; Scott, M.P. Conservation of the hedgehog/patched signaling pathway from flies to mice: Induction of a mouse patched gene by Hedgehog. Genes Dev. 1996, 10, 301–312. [Google Scholar] [CrossRef]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. Dual roles for patched in sequestering and transducing Hedgehog. Cell 1996, 87, 553–563. [Google Scholar] [CrossRef]
- Quirk, J.; van den Heuvel, M.; Henrique, D.; Marigo, V.; Jones, T.A.; Tabin, C.; Ingham, P.W. The smoothened gene and hedgehog signal transduction in Drosophila and vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 1997, 62, 217–226. [Google Scholar]
- Denef, N.; Neubuser, D.; Perez, L.; Cohen, S.M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000, 102, 521–531. [Google Scholar] [CrossRef]
- Ingham, P.W.; Nystedt, S.; Nakano, Y.; Brown, W.; Stark, D.; van den Heuvel, M.; Taylor, A.M. Patched represses the Hedgehog signalling pathway by promoting modification of the Smoothened protein. Curr. Biol. 2000, 10, 1315–1318. [Google Scholar] [CrossRef]
- Casali, A.; Struhl, G. Reading the Hedgehog morphogen gradient by measuring the ratio of bound to unbound Patched protein. Nature 2004, 431, 76–80. [Google Scholar] [CrossRef]
- Incardona, J.P.; Lee, J.H.; Robertson, C.P.; Enga, K.; Kapur, R.P.; Roelink, H. Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched-1. Proc. Natl. Acad. Sci. USA 2000, 97, 12044–12049. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H., Jr. The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar]
- Corcoran, R.B.; Scott, M.P. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8408–8413. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e1114. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef]
- Sommer, A.; Lemmon, M.A. Smoothening out the patches. Science 2018, 362, 26–27. [Google Scholar] [CrossRef]
- Eguether, T.; Hahne, M. Mixed signals from the cell’s antennae: Primary cilia in cancer. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Zaghloul, N.A.; Brugmann, S.A. The emerging face of primary cilia. Genesis 2011, 49, 231–246. [Google Scholar] [CrossRef]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Pearse, R.V., 2nd; Collier, L.S.; Scott, M.P.; Tabin, C.J. Vertebrate homologs of Drosophila suppressor of fused interact with the gli family of transcriptional regulators. Dev. Biol. 1999, 212, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.M.; Murone, M.; Luoh, S.; Ye, W.; Armanini, M.P.; Gurney, A.; Phillips, H.; Brush, J.; Goddard, A.; de Sauvage, F.J.; et al. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J. Cell Sci. 1999, 112 Pt 23, 4437–4448. [Google Scholar]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear–cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312. [Google Scholar] [CrossRef] [PubMed]
- Tuson, M.; He, M.; Anderson, K.V. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J. Craniofacial Ciliopathies and the Interpretation of Hedgehog Signal Transduction. Plos Genet. 2016, 12, e1006460. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Couly, G.; Le Douarin, N.M. Head morphogenesis in embryonic avian chimeras: Evidence for a segmental pattern in the ectoderm corresponding to the neuromeres. Development 1990, 108, 543–558. [Google Scholar]
- Noden, D.M. Interactions and fates of avian craniofacial mesenchyme. Development 1988, 103, 121–140. [Google Scholar] [PubMed]
- Osumi-Yamashita, N.; Ninomiya, Y.; Doi, H.; Eto, K. The contribution of both forebrain and midbrain crest cells to the mesenchyme in the frontonasal mass of mouse embryos. Dev. Biol. 1994, 164, 409–419. [Google Scholar] [CrossRef]
- Cerny, R.; Lwigale, P.; Ericsson, R.; Meulemans, D.; Epperlein, H.H.; Bronner-Fraser, M. Developmental origins and evolution of jaws: New interpretation of “maxillary” and “mandibular”. Dev. Biol. 2004, 276, 225–236. [Google Scholar] [CrossRef]
- Helms, J.A.; Cordero, D.; Tapadia, M.D. New insights into craniofacial morphogenesis. Development 2005, 132, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Abramyan, J.; Thivichon-Prince, B.; Richman, J.M. Diversity in primary palate ontogeny of amniotes revealed with 3D imaging. J. Anat. 2015, 226, 420–433. [Google Scholar] [CrossRef]
- Ahlgren, S.C.; Thakur, V.; Bronner-Fraser, M. Sonic hedgehog rescues cranial neural crest from cell death induced by ethanol exposure. Proc. Natl. Acad. Sci. USA 2002, 99, 10476–10481. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Mao, J.; Tenzen, T.; Kottmann, A.H.; McMahon, A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004, 18, 937–951. [Google Scholar] [CrossRef]
- Couly, G.; Creuzet, S.; Bennaceur, S.; Vincent, C.; Le Douarin, N.M. Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development 2002, 129, 1061–1073. [Google Scholar] [PubMed]
- Benouaiche, L.; Gitton, Y.; Vincent, C.; Couly, G.; Levi, G. Sonic hedgehog signalling from foregut endoderm patterns the avian nasal capsule. Development 2008, 135, 2221–2225. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.M.; Teillet, M.A.; Le Douarin, N.M. An early role for sonic hedgehog from foregut endoderm in jaw development: Ensuring neural crest cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 11607–11612. [Google Scholar] [CrossRef]
- Chen, W.; Burgess, S.; Hopkins, N. Analysis of the zebrafish smoothened mutant reveals conserved and divergent functions of hedgehog activity. Development 2001, 128, 2385–2396. [Google Scholar] [PubMed]
- Bolande, R.P. The neurocristopathies: A unifying concept of disease arising in neural crest maldevelopment. Hum. Pathol. 1974, 5, 409–429. [Google Scholar] [CrossRef]
- Hall, B.K. The Neural Crest and Neural Crest Cells in Vertebrate Development and Evolution; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008; Volume 11. [Google Scholar]
- Hans, J.; Lammens, M.; Hori, A. Clinical Neuroembryology: Development and Developmental Disorders of the Human Central Nervous System; Springer Science+Business Media: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Vega-Lopez, G.A.; Cerrizuela, S.; Tribulo, C.; Aybar, M.J. Neurocristopathies: New insights 150 years after the neural crest discovery. Dev. Biol. 2018, 444 (Suppl. 1), S110–S143. [Google Scholar] [CrossRef]
- Kurosaka, H.; Trainor, P.A.; Leroux-Berger, M.; Iulianella, A. Cranial nerve development requires co-ordinated Shh and canonical Wnt signaling. PLoS ONE 2015, 10, e0120821. [Google Scholar] [CrossRef] [PubMed]
- Currier, D.G.; Polk, R.C.; Reeves, R.H. A Sonic hedgehog (Shh) response deficit in trisomic cells may be a common denominator for multiple features of Down syndrome. Prog. Brain Res. 2012, 197, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Roper, R.J.; VanHorn, J.F.; Cain, C.C.; Reeves, R.H. A neural crest deficit in Down syndrome mice is associated with deficient mitotic response to Sonic hedgehog. Mech. Dev. 2009, 126, 212–219. [Google Scholar] [CrossRef]
- Richman, J.M.; Tickle, C. Epithelia are interchangeable between facial primordia of chick embryos and morphogenesis is controlled by the mesenchyme. Dev. Biol. 1989, 136, 201–210. [Google Scholar] [CrossRef]
- Hu, D.; Helms, J.A. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development 1999, 126, 4873–4884. [Google Scholar]
- Hu, D.; Marcucio, R.S.; Helms, J.A. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development 2003, 130, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Abzhanov, A.; Tabin, C.J. Shh and Fgf8 act synergistically to drive cartilage outgrowth during cranial development. Dev. Biol. 2004, 273, 134–148. [Google Scholar] [CrossRef]
- Marcucio, R.S.; Cordero, D.R.; Hu, D.; Helms, J.A. Molecular interactions coordinating the development of the forebrain and face. Dev. Biol. 2005, 284, 48–61. [Google Scholar] [CrossRef]
- Ming, J.E.; Roessler, E.; Muenke, M. Human developmental disorders and the Sonic hedgehog pathway. Mol. Med. Today 1998, 4, 343–349. [Google Scholar] [CrossRef]
- Lupo, G.; Harris, W.A.; Lewis, K.E. Mechanisms of ventral patterning in the vertebrate nervous system. Nat. Rev. Neurosci. 2006, 7, 103–114. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Costa, M.A.; Borzabadi-Farahani, A.; Lara-Sanchez, P.A.; Schweitzer, D.; Jacobson, L.; Clarke, N.; Hammoudeh, J.; Urata, M.M.; Magee, W.P., 3rd. Partial craniofacial duplication: A review of the literature and case report. J. Cranio-Maxillo-Facial Surg. 2014, 42, 290–296. [Google Scholar] [CrossRef]
- Croen, L.A.; Shaw, G.M.; Lammer, E.J. Holoprosencephaly: Epidemiologic and clinical characteristics of a California population. Am. J. Med. Genet. 1996, 64, 465–472. [Google Scholar] [CrossRef]
- Bale, A.E. Hedgehog signaling and human disease. Annu. Rev. Genom. Hum. Genet. 2002, 3, 47–65. [Google Scholar] [CrossRef]
- Wilkie, A.O. Craniosynostosis: Genes and mechanisms. Hum. Mol. Genet. 1997, 6, 1647–1656. [Google Scholar] [CrossRef]
- Aoto, K.; Shikata, Y.; Imai, H.; Matsumaru, D.; Tokunaga, T.; Shioda, S.; Yamada, G.; Motoyama, J. Mouse Shh is required for prechordal plate maintenance during brain and craniofacial morphogenesis. Dev. Biol. 2009, 327, 106–120. [Google Scholar] [CrossRef]
- Muenke, M.; Beachy, P.A. Genetics of ventral forebrain development and holoprosencephaly. Curr. Opin. Genet. Dev. 2000, 10, 262–269. [Google Scholar] [CrossRef]
- Aoto, K.; Trainor, P.A. Co-ordinated brain and craniofacial development depend upon Patched1/XIAP regulation of cell survival. Hum. Mol. Genet. 2015, 24, 698–713. [Google Scholar] [CrossRef]
- Dennis, J.F.; Kurosaka, H.; Iulianella, A.; Pace, J.; Thomas, N.; Beckham, S.; Williams, T.; Trainor, P.A. Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PLoS Genet. 2012, 8, e1002927. [Google Scholar] [CrossRef]
- Geng, X.; Oliver, G. Pathogenesis of holoprosencephaly. J. Clin. Investig. 2009, 119, 1403–1413. [Google Scholar] [CrossRef]
- Winter, T.C.; Kennedy, A.M.; Woodward, P.J. Holoprosencephaly: A survey of the entity, with embryology and fetal imaging. Radiographics 2015, 35, 275–290. [Google Scholar] [CrossRef]
- Nieuwenhuis, E.; Hui, C.C. Hedgehog signaling and congenital malformations. Clin. Genet. 2005, 67, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.E.; Muenke, M. Multiple hits during early embryonic development: Digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 2002, 71, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Beachy, P.A. Patterning of the embryonic forebrain. Curr. Opin. Neurobiol. 1998, 8, 18–26. [Google Scholar] [CrossRef]
- Ming, J.E.; Muenke, M. Holoprosencephaly: From Homer to Hedgehog. Clin. Genet. 1998, 53, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Mercier, S.; Dubourg, C.; Garcelon, N.; Campillo-Gimenez, B.; Gicquel, I.; Belleguic, M.; Ratie, L.; Pasquier, L.; Loget, P.; Bendavid, C.; et al. New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J. Med. Genet. 2011, 48, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Leskow, F.C.; El-Jaick, K.; Roessler, E.; Muenke, M.; Yocum, A.; Dubourg, C.; Li, X.; Geng, X.; Oliver, G.; et al. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 2008, 40, 1348–1353. [Google Scholar] [CrossRef]
- Warr, N.; Powles-Glover, N.; Chappell, A.; Robson, J.; Norris, D.; Arkell, R.M. Zic2-associated holoprosencephaly is caused by a transient defect in the organizer region during gastrulation. Hum. Mol. Genet. 2008, 17, 2986–2996. [Google Scholar] [CrossRef]
- Seppala, M.; Depew, M.J.; Martinelli, D.C.; Fan, C.M.; Sharpe, P.T.; Cobourne, M.T. Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J. Clin. Investig. 2007, 117, 1575–1584. [Google Scholar] [CrossRef]
- Cole, F.; Krauss, R.S. Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Curr. Biol. 2003, 13, 411–415. [Google Scholar] [CrossRef]
- Okada, A.; Charron, F.; Morin, S.; Shin, D.S.; Wong, K.; Fabre, P.J.; Tessier-Lavigne, M.; McConnell, S.K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 2006, 444, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Du, Y.Z.; Mullor, J.L.; Casas, E.; Allen, W.P.; Gillessen-Kaesbach, G.; Roeder, E.R.; Ming, J.E.; Ruiz i Altaba, A.; Muenke, M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. USA 2003, 100, 13424–13429. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Hilpert, J.; Armstrong, S.A.; Rohlmann, A.; Hammer, R.E.; Burns, D.K.; Herz, J. Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. USA 1996, 93, 8460–8464. [Google Scholar] [CrossRef] [PubMed]
- Izraeli, S.; Lowe, L.A.; Bertness, V.L.; Campaner, S.; Hahn, H.; Kirsch, I.R.; Kuehn, M.R. Genetic evidence that Sil is required for the Sonic Hedgehog response pathway. Genesis 2001, 31, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 2001, 105, 781–792. [Google Scholar] [CrossRef]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Muenke, M. Holoprosencephaly: A paradigm for the complex genetics of brain development. J. Inherit. Metab. Dis. 1998, 21, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.L.; Roessler, E.; Hennekam, R.C.; Feldman, G.L.; Kosaki, K.; Jones, M.C.; Palumbos, J.C.; Muenke, M. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: Does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am. J. Med. Genet. 1996, 66, 478–484. [Google Scholar] [CrossRef]
- Abramyan, J.; Leung, K.J.; Richman, J.M. Divergent palate morphology in turtles and birds correlates with differences in proliferation and BMP2 expression during embryonic development. J. Exp. Zool. Part B Mol. Dev. Evol. 2014, 322, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Abramyan, J.; Richman, J.M. Recent insights into the morphological diversity in the amniote primary and secondary palates. Dev. Dyn. 2015, 244, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Heyne, G.W.; Melberg, C.G.; Doroodchi, P.; Parins, K.F.; Kietzman, H.W.; Everson, J.L.; Ansen-Wilson, L.J.; Lipinski, R.J. Definition of critical periods for Hedgehog pathway antagonist-induced holoprosencephaly, cleft lip, and cleft palate. PLoS ONE 2015, 10, e0120517. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Song, C.; Sulik, K.K.; Everson, J.L.; Gipp, J.J.; Yan, D.; Bushman, W.; Rowland, I.J. Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: Phenotypic characterization and clinical implications. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.A.; Kim, C.H.; Hu, D.; Minkoff, R.; Thaller, C.; Eichele, G. Sonic hedgehog participates in craniofacial morphogenesis and is down-regulated by teratogenic doses of retinoic acid. Dev. Biol. 1997, 187, 25–35. [Google Scholar] [CrossRef]
- Young, D.L.; Schneider, R.A.; Hu, D.; Helms, J.A. Genetic and teratogenic approaches to craniofacial development. Crit. Rev. Oral Biol. Med. 2000, 11, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Metzis, V.; Courtney, A.D.; Kerr, M.C.; Ferguson, C.; Rondon Galeano, M.C.; Parton, R.G.; Wainwright, B.J.; Wicking, C. Patched1 is required in neural crest cells for the prevention of orofacial clefts. Hum. Mol. Genet. 2013, 22, 5026–5035. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, Y.; He, J.; Feng, J.; Han, X.; Jing, J.; Ho, T.V.; Xu, J.; Chai, Y. Constitutive activation of hedgehog signaling adversely affects epithelial cell fate during palatal fusion. Dev. Biol. 2018, 441, 191–203. [Google Scholar] [CrossRef]
- Cobourne, M.T.; Xavier, G.M.; Depew, M.; Hagan, L.; Sealby, J.; Webster, Z.; Sharpe, P.T. Sonic hedgehog signalling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Dev. Biol. 2009, 331, 38–49. [Google Scholar] [CrossRef]
- Hammond, N.L.; Brookes, K.J.; Dixon, M.J. Ectopic Hedgehog Signaling Causes Cleft Palate and Defective Osteogenesis. J. Dent. Res. 2018, 97, 1485–1493. [Google Scholar] [CrossRef]
- Kurosaka, H.; Iulianella, A.; Williams, T.; Trainor, P.A. Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. J. Clin. Investig. 2014, 124, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Brugmann, S.A.; Cordero, D.R.; Helms, J.A. Craniofacial ciliopathies: A new classification for craniofacial disorders. Am. J. Med. Genet. Part A 2010, 152a, 2995–3006. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Briscoe, J. Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 753–772. [Google Scholar] [CrossRef]
- Avasthi, P.; Marshall, W.F. Stages of ciliogenesis and regulation of ciliary length. Differentiation 2012, 83, S30–S42. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Liu, A.; Wang, B.; Niswander, L.A. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 2005, 132, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genom. Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Beales, P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151c, 281–295. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatric Nephrol. (Berl. Ger.) 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Cortes, C.R.; Metzis, V.; Wicking, C. Unmasking the ciliopathies: Craniofacial defects and the primary cilium. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 637–653. [Google Scholar] [CrossRef]
- Brugmann, S.A.; Allen, N.C.; James, A.W.; Mekonnen, Z.; Madan, E.; Helms, J.A. A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet. 2010, 19, 1577–1592. [Google Scholar] [CrossRef]
- Cordero, D.; Marcucio, R.; Hu, D.; Gaffield, W.; Tapadia, M.; Helms, J.A. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J. Clin. Investig. 2004, 114, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Witman, G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Scholey, J.M.; Anderson, K.V. Intraflagellar transport and cilium-based signaling. Cell 2006, 125, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Ocbina, P.J.; Anderson, K.V. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: Analysis in mouse embryonic fibroblasts. Dev. Dyn. 2008, 237, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Anderson, K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 11325–11330. [Google Scholar] [CrossRef]
- Haycraft, C.J.; Zhang, Q.; Song, B.; Jackson, W.S.; Detloff, P.J.; Serra, R.; Yoder, B.K. Intraflagellar transport is essential for endochondral bone formation. Development 2007, 134, 307–316. [Google Scholar] [CrossRef]
- Keady, B.T.; Samtani, R.; Tobita, K.; Tsuchya, M.; San Agustin, J.T.; Follit, J.A.; Jonassen, J.A.; Subramanian, R.; Lo, C.W.; Pazour, G.J. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell 2012, 22, 940–951. [Google Scholar] [CrossRef]
- May, S.R.; Ashique, A.M.; Karlen, M.; Wang, B.; Shen, Y.; Zarbalis, K.; Reiter, J.; Ericson, J.; Peterson, A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005, 287, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Wang, C.; Wang, B.; Bassi, M.R.; Caretti, E.; Champeval, D.; Calmont, A.; Jarnik, M.; Burch, J.; Zaret, K.S.; et al. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 2009, 325, 225–237. [Google Scholar] [CrossRef]
- Tran, P.V.; Haycraft, C.J.; Besschetnova, T.Y.; Turbe-Doan, A.; Stottmann, R.W.; Herron, B.J.; Chesebro, A.L.; Qiu, H.; Scherz, P.J.; Shah, J.V.; et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008, 40, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Schock, E.N.; Chang, C.F.; Youngworth, I.A.; Davey, M.G.; Delany, M.E.; Brugmann, S.A. Utilizing the chicken as an animal model for human craniofacial ciliopathies. Dev. Biol. 2016, 415, 326–337. [Google Scholar] [CrossRef]
- Cole, R.K. The “talpid lethal” in the domestic fowl. J. Hered. 1942, 33, 83–86. [Google Scholar] [CrossRef]
- Chang, C.F.; Schock, E.N.; O’Hare, E.A.; Dodgson, J.; Cheng, H.H.; Muir, W.M.; Edelmann, R.E.; Delany, M.E.; Brugmann, S.A. The cellular and molecular etiology of the craniofacial defects in the avian ciliopathic mutant talpid2. Development 2014, 141, 3003–3012. [Google Scholar] [CrossRef]
- Hoover, A.N.; Wynkoop, A.; Zeng, H.; Jia, J.; Niswander, L.A.; Liu, A. C2cd3 is required for cilia formation and Hedgehog signaling in mouse. Development 2008, 135, 4049–4058. [Google Scholar] [CrossRef]
- Thauvin-Robinet, C.; Lee, J.S.; Lopez, E.; Herranz-Perez, V.; Shida, T.; Franco, B.; Jego, L.; Ye, F.; Pasquier, L.; Loget, P.; et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat. Genet. 2014, 46, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zeng, H.; Ning, G.; Reiter, J.F.; Liu, A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 2164–2169. [Google Scholar] [CrossRef]
- Agarwala, S.; Aglyamova, G.V.; Marma, A.K.; Fallon, J.F.; Ragsdale, C.W. Differential susceptibility of midbrain and spinal cord patterning to floor plate defects in the talpid2 mutant. Dev. Biol. 2005, 288, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.P.; Hasso, S.M.; Ferguson, M.W.; Fallon, J.F. The development of archosaurian first-generation teeth in a chicken mutant. Curr. Biol. 2006, 16, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.A.; Hu, D.; Helms, J.A. From head to toe: Conservation of molecular signals regulating limb and craniofacial morphogenesis. Cell Tissue Res. 1999, 296, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ede, D.A.; Kelly, W.A. Developmental abnormalities in the head region of the talpid3 mutant of the fowl. Development 1964, 12, 161–182. [Google Scholar]
- Ede, D.A.; Kelly, W.A. Developmental abnormalities in the trunk and limbs of the talpid3 mutant of the fowl. Development 1964, 12, 339–356. [Google Scholar]
- Davey, M.G.; Paton, I.R.; Yin, Y.; Schmidt, M.; Bangs, F.K.; Morrice, D.R.; Smith, T.G.; Buxton, P.; Stamataki, D.; Tanaka, M.; et al. The chicken talpid3 gene encodes a novel protein essential for Hedgehog signaling. Genes Dev. 2006, 20, 1365–1377. [Google Scholar] [CrossRef]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kim, S.; Lin, Y.C.; Inoue, T.; Dynlacht, B.D. The CP110-interacting proteins Talpid3 and Cep290 play overlapping and distinct roles in cilia assembly. J. Cell Biol. 2014, 204, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Antonio, N.; Thongnuek, P.; Welten, M.; Davey, M.G.; Briscoe, J.; Tickle, C. Generation of mice with functional inactivation of talpid3, a gene first identified in chicken. Development 2011, 138, 3261–3272. [Google Scholar] [CrossRef]
- Ben, J.; Elworthy, S.; Ng, A.S.; van Eeden, F.; Ingham, P.W. Targeted mutation of the talpid3 gene in zebrafish reveals its conserved requirement for ciliogenesis and Hedgehog signalling across the vertebrates. Development 2011, 138, 4969–4978. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.A.; Davis, G.M.; McTeir, K.E.; James, J.; McTeir, L.; Kierans, M.; Bain, A.; Davey, M.G. Failure of centrosome migration causes a loss of motile cilia in talpid(3) mutants. Dev. Dyn. 2013, 242, 923–931. [Google Scholar] [CrossRef]
- Yin, Y.; Bangs, F.; Paton, I.R.; Prescott, A.; James, J.; Davey, M.G.; Whitley, P.; Genikhovich, G.; Technau, U.; Burt, D.W.; et al. The Talpid3 gene (KIAA0586) encodes a centrosomal protein that is essential for primary cilia formation. Development 2009, 136, 655–664. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Phelps, I.G.; Dempsey, J.C.; Sharma, V.A.; Ishak, G.E.; Boyle, E.A.; Wilson, M.; Marques Lourenco, C.; Arslan, M.; Shendure, J.; et al. KIAA0586 is Mutated in Joubert Syndrome. Hum. Mutat. 2015, 36, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Hofree, M.; Kim, S.; Scott, E.; Copeland, B.; Romani, M.; Silhavy, J.L.; Rosti, R.O.; Schroth, J.; Mazza, T.; et al. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. eLife 2015, 4, e06602. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nurnberg, P.; Nurnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). eLife 2015, 4. [Google Scholar] [CrossRef]
- Buxton, P.; Davey, M.G.; Paton, I.R.; Morrice, D.R.; Francis-West, P.H.; Burt, D.W.; Tickle, C. Craniofacial development in the talpid3 chicken mutant. Differentiation 2004, 72, 348–362. [Google Scholar] [CrossRef]
- Matsubara, Y.; Nakano, M.; Kawamura, K.; Tsudzuki, M.; Funahashi, J.I.; Agata, K.; Matsuda, Y.; Kuroiwa, A.; Suzuki, T. Inactivation of Sonic Hedgehog Signaling and Polydactyly in Limbs of Hereditary Multiple Malformation, a Novel Type of Talpid Mutant. Front. Cell Dev. Biol. 2016, 4, 149. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001. [Google Scholar] [CrossRef]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rehm, J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e290–e299. [Google Scholar] [CrossRef]
- Calhoun, F.; Warren, K. Fetal alcohol syndrome: Historical perspectives. Neurosci. Biobehav. Rev. 2007, 31, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L. The effects of alcohol on fetal development. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Siebert, J.R.; Astley, S.J.; Clarren, S.K. Holoprosencephaly in a fetal macaque (Macaca nemestrina) following weekly exposure to ethanol. Teratology 1991, 44, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Charness, M.E.; Safran, R.M.; Perides, G. Ethanol inhibits neural cell-cell adhesion. J. Biol. Chem. 1994, 269, 9304–9309. [Google Scholar]
- Kotch, L.E.; Chen, S.Y.; Sulik, K.K. Ethanol-induced teratogenesis: Free radical damage as a possible mechanism. Teratology 1995, 52, 128–136. [Google Scholar] [CrossRef]
- Henderson, G.I.; Baskin, G.S.; Horbach, J.; Porter, P.; Schenker, S. Arrest of epidermal growth factor-dependent growth in fetal hepatocytes after ethanol exposure. J. Clin. Investig. 1989, 84, 1287–1294. [Google Scholar] [CrossRef]
- Deltour, L.; Ang, H.L.; Duester, G. Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J. 1996, 10, 1050–1057. [Google Scholar] [CrossRef]
- Kietzman, H.W.; Everson, J.L.; Sulik, K.K.; Lipinski, R.J. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS ONE 2014, 9, e89448. [Google Scholar] [CrossRef]
- Sulik, K.K.; Johnston, M.C. Sequence of developmental alterations following acute ethanol exposure in mice: Craniofacial features of the fetal alcohol syndrome. Am. J. Anat. 1983, 166, 257–269. [Google Scholar] [CrossRef]
- Bupp Becker, S.R.; Shibley, I.A., Jr. Teratogenicity of ethanol in different chicken strains. Alcohol Alcohol. 1998, 33, 457–464. [Google Scholar] [CrossRef]
- Bilotta, J.; Barnett, J.A.; Hancock, L.; Saszik, S. Ethanol exposure alters zebrafish development: A novel model of fetal alcohol syndrome. Neurotoxicol. Teratol. 2004, 26, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Yang, H.T.; Zdanowicz, M.; Sicklick, J.K.; Qi, Y.; Camp, T.J.; Diehl, A.M. Fetal alcohol exposure impairs Hedgehog cholesterol modification and signaling. Lab. Investig. A J. Tech. Methods Pathol. 2007, 87, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Shikata, Y.; Higashiyama, D.; Shiota, K.; Motoyama, J. Fetal ethanol exposure activates protein kinase A and impairs Shh expression in prechordal mesendoderm cells in the pathogenesis of holoprosencephaly. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Rovasio, R.A.; Battiato, N.L. Role of early migratory neural crest cells in developmental anomalies induced by ethanol. Int. J. Dev. Biol. 1995, 39, 421–422. [Google Scholar]
- Rovasio, R.A.; Battiato, N.L. Ethanol induces morphological and dynamic changes on in vivo and in vitro neural crest cells. Alcohol. Clin. Exp. Res. 2002, 26, 1286–1298. [Google Scholar] [CrossRef]
- Brennan, D.; Giles, S. Sonic hedgehog expression is disrupted following in ovo ethanol exposure during early chick eye development. Reprod. Toxicol. 2013, 41, 49–56. [Google Scholar] [CrossRef]
- Tolosa, E.J.; Fernandez-Zapico, M.E.; Battiato, N.L.; Rovasio, R.A. Sonic hedgehog is a chemotactic neural crest cell guide that is perturbed by ethanol exposure. Eur. J. Cell Biol. 2016, 95, 136–152. [Google Scholar] [CrossRef]
- Streissguth, A.P.; Dehaene, P. Fetal alcohol syndrome in twins of alcoholic mothers: Concordance of diagnosis and IQ. Am. J. Med. Genet. 1993, 47, 857–861. [Google Scholar] [CrossRef]
- Chasnoff, I.J. Fetal alcohol syndrome in twin pregnancy. Acta Genet. Med. Gemellol. 1985, 34, 229–232. [Google Scholar] [CrossRef]
- Ding, Q.; Motoyama, J.; Gasca, S.; Mo, R.; Sasaki, H.; Rossant, J.; Hui, C.C. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 1998, 125, 2533–2543. [Google Scholar]
- Mo, R.; Freer, A.M.; Zinyk, D.L.; Crackower, M.A.; Michaud, J.; Heng, H.H.; Chik, K.W.; Shi, X.M.; Tsui, L.C.; Cheng, S.H.; et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 1997, 124, 113–123. [Google Scholar]
- Hong, M.; Krauss, R.S. Cdon mutation and fetal ethanol exposure synergize to produce midline signaling defects and holoprosencephaly spectrum disorders in mice. PLoS Genet. 2012, 8, e1002999. [Google Scholar] [CrossRef]
- Hong, M.; Krauss, R.S. Rescue of holoprosencephaly in fetal alcohol-exposed Cdon mutant mice by reduced gene dosage of Ptch1. PLoS ONE 2013, 8, e79269. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Wassif, C.A.; Krakowiak, P.A.; Taipale, J.; Gong, R.; Kelley, R.I.; Porter, F.D.; Beachy, P.A. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 2003, 33, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Edison, R.J.; Muenke, M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N. Engl. J. Med. 2004, 350, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Maslen, C.; Kachilele-Linjewile, S.; Lin, D.; Linck, L.M.; Connor, W.E.; Steiner, R.D.; Porter, F.D. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am. J. Hum. Genet. 1998, 63, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Horvath, C.; Dupuis, R. Teratogenic action and embryo lethality of AY 9944R. Prevention by a hypercholesterolemia-provoking diet. Teratology 1979, 19, 35–38. [Google Scholar] [CrossRef]
- Napoli, C.; D’Armiento, F.P.; Mancini, F.P.; Postiglione, A.; Witztum, J.L.; Palumbo, G.; Palinski, W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Glass, C.K.; Witztum, J.L.; Deutsch, R.; D’Armiento, F.P.; Palinski, W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet 1999, 354, 1234–1241. [Google Scholar] [CrossRef]
- Palinski, W.; D’Armiento, F.P.; Witztum, J.L.; de Nigris, F.; Casanada, F.; Condorelli, M.; Silvestre, M.; Napoli, C. Maternal hypercholesterolemia and treatment during pregnancy influence the long-term progression of atherosclerosis in offspring of rabbits. Circ. Res. 2001, 89, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Iseki, S.; Maxson, R.E.; Sucov, H.M.; Morriss-Kay, G.M. Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 2002, 241, 106–116. [Google Scholar] [CrossRef]
- Chai, Y.; Maxson, R.E., Jr. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006, 235, 2353–2375. [Google Scholar] [CrossRef] [PubMed]
- Opperman, L.A. Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 2000, 219, 472–485. [Google Scholar] [CrossRef]
- Lenton, K.A.; Nacamuli, R.P.; Wan, D.C.; Helms, J.A.; Longaker, M.T. Cranial suture biology. Curr. Top. Dev. Biol. 2005, 66, 287–328. [Google Scholar] [CrossRef]
- Bristol, R.E.; Lekovic, G.P.; Rekate, H.L. The effects of craniosynostosis on the brain with respect to intracranial pressure. Semin. Pediatr. Neurol. 2004, 11, 262–267. [Google Scholar] [CrossRef]
- Kapp-Simon, K.A. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate-Craniofacial J. 1998, 35, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Panchal, J.; Amirsheybani, H.; Gurwitch, R.; Cook, V.; Francel, P.; Neas, B.; Levine, N. Neurodevelopment in children with single-suture craniosynostosis and plagiocephaly without synostosis. Plast. Reconstr. Surg. 2001, 108, 1492–1498, discussion 1499-1500. [Google Scholar] [CrossRef]
- Macintosh, C.; Wells, R.; Johnson, D.; Wall, S. What are the effects of metopic synostosis on visual function? J. Craniofac. Surg. 2011, 22, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Chang, L.; Nguyen, A.; James, A.W. A review of hedgehog signaling in cranial bone development. Front. Physiol. 2013, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Nott, R.L.; Stelnicki, E.J.; Mack, J.A.; Ben, Y.; Mitchell, R.; Mooney, M.P. Comparison of hedgehog and patched-1 protein expression in the cranial sutures of craniosynostotic and wild-type rabbits. Plast. Reconstr. Surg. 2002, 110, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Klopocki, E.; Lohan, S.; Brancati, F.; Koll, R.; Brehm, A.; Seemann, P.; Dathe, K.; Stricker, S.; Hecht, J.; Bosse, K.; et al. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am. J. Hum. Genet. 2011, 88, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Nifuji, A.; Noda, M. Expression of Indian hedgehog in osteoblasts and its posttranscriptional regulation by transforming growth factor-beta. Endocrinology 1997, 138, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Veistinen, L.K.; Mustonen, T.; Hasan, M.R.; Takatalo, M.; Kobayashi, Y.; Kesper, D.A.; Vortkamp, A.; Rice, D.P. Regulation of Calvarial Osteogenesis by Concomitant De-repression of GLI3 and Activation of IHH Targets. Front. Physiol. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Wu, C.; Freeman, T.A.; Koyama, E.; Kirschner, R.E. Expression of Indian Hedgehog, BMP-4 and Noggin in craniosynostosis induced by fetal constraint. Ann. Plast. Surg. 2007, 58, 215–221. [Google Scholar] [CrossRef]
- Lenton, K.; James, A.W.; Manu, A.; Brugmann, S.A.; Birker, D.; Nelson, E.R.; Leucht, P.; Helms, J.A.; Longaker, M.T. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis 2011, 49, 784–796. [Google Scholar] [CrossRef]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar] [CrossRef]
- Kim, H.J.; Rice, D.P.; Kettunen, P.J.; Thesleff, I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development 1998, 125, 1241–1251. [Google Scholar]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abramyan, J. Hedgehog Signaling and Embryonic Craniofacial Disorders. J. Dev. Biol. 2019, 7, 9. https://doi.org/10.3390/jdb7020009
Abramyan J. Hedgehog Signaling and Embryonic Craniofacial Disorders. Journal of Developmental Biology. 2019; 7(2):9. https://doi.org/10.3390/jdb7020009
Chicago/Turabian StyleAbramyan, John. 2019. "Hedgehog Signaling and Embryonic Craniofacial Disorders" Journal of Developmental Biology 7, no. 2: 9. https://doi.org/10.3390/jdb7020009
APA StyleAbramyan, J. (2019). Hedgehog Signaling and Embryonic Craniofacial Disorders. Journal of Developmental Biology, 7(2), 9. https://doi.org/10.3390/jdb7020009