Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model

Abstract
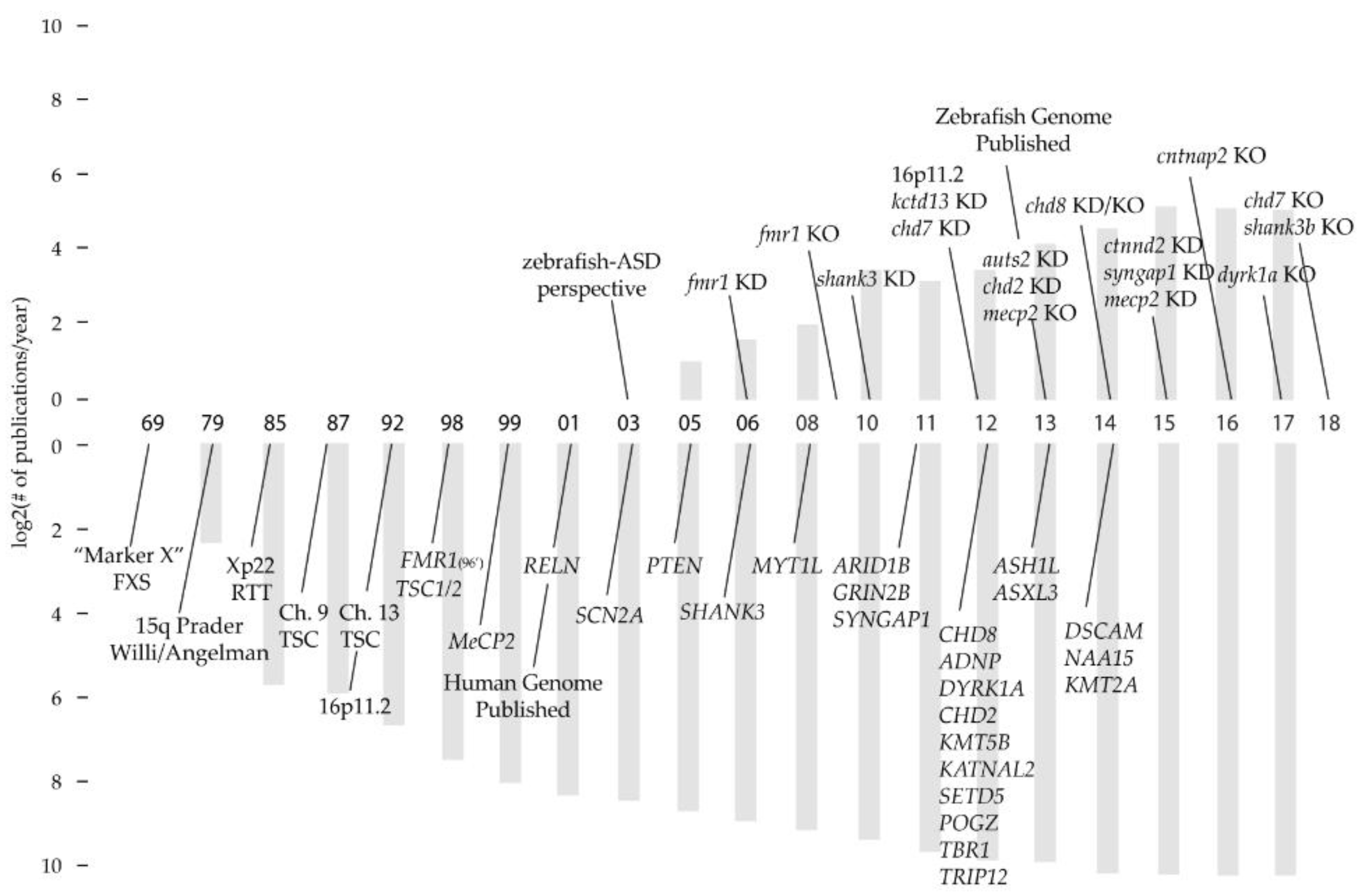
1. Introduction
2. What Prenatal Factors Contribute to ASD-ID
2.1. Environmental Risk Factors
2.2. Genetic Risk Factors
3. Comparative Neurodevelopment and Critical Periods: Can Embryonic and Larval Periods in Zebrafish be Compared to Prenatal Development in Humans?
3.1. Conserved Neuronal Development with Differences in Morphogenesis
3.2. Integrated Critical Periods: Gene Expression, Morphogenesis and Function
4. Zebrafish ASD-ID Models: Critical Periods and Developmental Mechanisms
4.1. Disruptions in Morphogenesis and Circuit Development of the Embryonic Brain
4.1.1. The Dysregulation of Morphogenic Genes and Conserved Signaling Pathways Influences Brain Size
4.1.2. Deficits in Neurogenesis Negatively Impact Axonogenesis and Circuit Connectivity
5. Current Limitations for Using Zebrafish to Study Human Disease and Disorder Genes
5.1. The Teleost Whole Genome Duplication
5.2. Gene Knockdowns Versus Knockouts
6. The Integrated Model: Utilizing Emerging Technology to Integrate Anatomy, Physiology and Behavior
6.1. Characteristics of Behaviors during Zebrafish Embryonic and Larval Development
6.2. Brain Atlases and High Resolution Morphometry
6.3. Whole-Brain Visual Outputs of Neuronal Activity
6.4. Small Molecule Screening and Drug Discovery
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Association, A.P. Intellectual Disability. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; DSM: Heerlen, The Netherlands, 2013. [Google Scholar]
- Fombonne, E. Epidemiological surveys of autism and other pervasive developmental disorders: An update. J. Autism. Dev. Disord. 2003, 33, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.L.; Shoemaker, M. Intellectual disability and its relationship to autism spectrum disorders. Res. Dev. Disabil. 2009, 30, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Betancur, C.; Xu, H.; Buxbaum, J.D.; Ma’ayan, A. Network- and attribute-based classifiers can prioritize genes and pathways for autism spectrum disorders and intellectual disability. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveillance Summer 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Geschwind, D.H. Autism genetics: Searching for specificity and convergence. Genome Biol. 2012, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feng, G. Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol. 2012, 22, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Penagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. 2015, 10, 111–144. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Sano, Y.; de Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2012, 17, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Belinson, H.; Nakatani, J.; Babineau, B.A.; Birnbaum, R.Y.; Ellegood, J.; Bershteyn, M.; McEvilly, R.J.; Long, J.M.; Willert, K.; Klein, O.D.; et al. Prenatal beta-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry 2016, 21, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Abrams, A.J.; James, D.M.; Buglo, E.; Yan, Q.; Dallman, J.E. Function Over Form: Modeling Groups of Inherited Neurological Conditions in Zebrafish. Front. Mol. Neurosci. 2016, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Hatta, K.; Metcalfe, W.K. Early axonal contacts during development of an identified dendrite in the brain of the zebrafish. Neuron 1990, 4, 535–545. [Google Scholar] [CrossRef]
- Higashijima, S.; Masino, M.A.; Mandel, G.; Fetcho, J.R. Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J. Neurophysiol. 2003, 90, 3986–3997. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013, 10, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Satou, C.; Kimura, Y.; Hirata, H.; Suster, M.L.; Kawakami, K.; Higashijima, S. Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development 2013, 140, 3927–3931. [Google Scholar] [CrossRef] [PubMed]
- Marquart, G.D.; Tabor, K.M.; Brown, M.; Strykowski, J.L.; Varshney, G.K.; LaFave, M.C.; Mueller, T.; Burgess, S.M.; Higashijima, S.; Burgess, H.A. A 3D Searchable Database of Transgenic Zebrafish Gal4 and Cre Lines for Functional Neuroanatomy Studies. Front. Neural Circuits 2015, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Chen, T.W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderon, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef] [PubMed]
- Fosque, B.F.; Sun, Y.; Dana, H.; Yang, C.T.; Ohyama, T.; Tadross, M.R.; Patel, R.; Zlatic, M.; Kim, D.S.; Ahrens, M.B.; et al. Neural circuits. Labeling of active neural circuits in vivo with designed calcium integrators. Science 2015, 347, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.B.; Li, J.M.; Orger, M.B.; Robson, D.N.; Schier, A.F.; Engert, F.; Portugues, R. Brain-wide neuronal dynamics during motor adaptation in zebrafish. Nature 2012, 485, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Brustein, E.; Saint-Amant, L.; Buss, R.R.; Chong, M.; McDearmid, J.R.; Drapeau, P. Steps during the development of the zebrafish locomotor network. J. Physiol. Paris 2003, 97, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Fero, K.Y.T.; Burgess, H.A. The Behavioral Repertoire of Larval Zebrafish. In Zebrafish Models in Neurobehavioral Research; Humana Press: New York, NY, USA, 2011; pp. 249–291. [Google Scholar]
- Warp, E.; Agarwal, G.; Wyart, C.; Friedmann, D.; Oldfield, C.S.; Conner, A.; Del Bene, F.; Arrenberg, A.B.; Baier, H.; Isacoff, E.Y. Emergence of patterned activity in the developing zebrafish spinal cord. Curr. Biol. 2012, 22, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Orger, M.B.; de Polavieja, G.G. Zebrafish Behavior: Opportunities and Challenges. Annu. Rev. Neurosci. 2017, 40, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Arndt, T.L.; Stodgell, C.J.; Rodier, P.M. The teratology of autism. Int. J. Dev. Neurosci. 2005, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.T.; Stromland, K.; Ventura, L.; Johansson, M.; Bandim, J.M.; Gillberg, C. Autism associated with conditions characterized by developmental errors in early embryogenesis: A mini review. Int. J. Dev. Neurosci. 2005, 23, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.; Jaffe, A.E.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am. J. Psychiatry 2014, 171, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L. Microscopic neuroanatomic abnormalities in autism. Pediatrics 1991, 87, 791–796. [Google Scholar] [PubMed]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Chess, S.; Fernandez, P. Neurologic damage and behavior disorder in rubella children. Am. Ann. Deaf 1980, 125, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Turnpenny, P.; Quinn, A.; Glover, S.; Lloyd, D.J.; Montgomery, T.; Dean, J.C. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 2000, 37, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Stromland, K.; Nordin, V.; Miller, M.; Akerstrom, B.; Gillberg, C. Autism in thalidomide embryopathy: A population study. Dev. Med. Child. Neurol. 1994, 36, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Seung, H.; Kwon, K.J.; Ko, M.J.; Lee, E.J.; Oh, H.A.; Choi, C.S.; Kim, K.C.; Gonzales, E.L.; You, J.S.; et al. Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS ONE 2014, 9, e104927. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L.; et al. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicol. Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Baronio, D.; Puttonen, H.A.J.; Sundvik, M.; Semenova, S.; Lehtonen, E.; Panula, P. Embryonic exposure to valproic acid affects the histaminergic system and the social behaviour of adult zebrafish (Danio rerio). Br. J. Pharmacol. 2018, 175, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, P.; Goines, P.E.; Tancredi, D.J.; Ashwood, P.; Hansen, R.L.; Hertz-Picciotto, I.; Van de Water, J. Neonatal Cytokine Profiles Associated With Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 442–451. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, M.; Svoboda, O.; Weijts, B.G.; Espin-Palazon, R.; Sapp, V.; Pietri, T.; Bagnat, M.; Muotri, A.R.; Traver, D. Mecp2 regulates tnfa during zebrafish embryonic development and acute inflammation. Dis. Model Mech. 2017, 10, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Folstein, S.E.; Piven, J. Etiology of autism: Genetic influences. Pediatrics 1991, 87, 767–773. [Google Scholar] [PubMed]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Schoenfeld, B.P.; Bell, A.J.; Choi, C.H.; Bradley, M.P.; Hinchey, P.; Kollaros, M.; Park, J.H.; McBride, S.M.; Dockendorff, T.C. Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J. Neurosci. 2010, 30, 6782–6792. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szucs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Pepkowitz, S.H.; Kerfoot, C.; de Rosa, M.J.; Poukens, V.; Wienecke, R.; de Clue, J.E.; Vinters, H.V. Tuberous sclerosis in a 20-week gestation fetus: Immunohistochemical study. Acta Neuropathol. 1997, 94, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, P.; Chiriboga, L.; Mizuguchi, M.; Yee, H.; Miller, D.C.; Greco, M.A. Tuberous sclerosis in a 19-week fetus: Immunohistochemical and molecular study of hamartin and tuberin. Pediatr. Dev. Pathol. 2002, 5, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Bordarier, C.; Lellouch-Tubiana, A.; Robain, O. Cardiac rhabdomyoma and tuberous sclerosis in three fetuses: A neuropathological study. Brain Dev. 1994, 16, 467–471. [Google Scholar] [CrossRef]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.-H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Durak, O.; de Anda, F.C.; Singh, K.K.; Leussis, M.P.; Petryshen, T.L.; Sklar, P.; Tsai, L.-H. Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of beta-catenin. Mol. Psychiatry 2015, 20, 388–397. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef]
- De Rubeis, S.; Buxbaum, J.D. Genetics and genomics of autism spectrum disorder: Embracing complexity. Hum. Mol. Genet. 2015, 24, R24–R31. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Capanu, M.; De Rubeis, S.; McCallum, K.; Buxbaum, J.D. Identification of rare causal variants in sequence-based studies: Methods and applications to VPS13B, a gene involved in Cohen syndrome and autism. PLoS Genet. 2014, 10, e1004729. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Cristino, A.S.; Williams, S.M.; Hawi, Z.; An, J.Y.; Bellgrove, M.A.; Schwartz, C.E.; Costa Lda, F.; Claudianos, C. Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol. Psychiatry 2014, 19, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Penn, O.; Borenstein, E.; Eichler, E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015, 25, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B.; Wade, M.J. What are maternal effects (and what are they not)? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Guilmatre, A.; Sharp, A.J. Parent of origin effects. Clin. Genet. 2012, 81, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.; Anney, R.; Gallagher, L.; Heron, E.A. A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet. 2017, 25, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, E.J.; Spradling, A.C. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 2018, 361, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Kindler, S.; Kreienkamp, H.J. Dendritic mRNA targeting and translation. Adv. Exp. Med. Biol. 2012, 970, 285–305. [Google Scholar] [PubMed]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar] [PubMed]
- Clancy, B.; Darlington, R.B.; Finlay, B.L. Translating developmental time across mammalian species. Neuroscience 2001, 105, 7–17. [Google Scholar] [CrossRef]
- Vaillancourt, C.; Lafond, J. Human embryogenesis: Overview. Methods Mol. Biol. 2009, 550, 3–7. [Google Scholar] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef] [PubMed]
- Moens, C.B.; Prince, V.E. Constructing the hindbrain: Insights from the zebrafish. Dev. Dyn. 2002, 224, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hibi, M. Development and evolution of cerebellar neural circuits. Dev. Growth Differ. 2012, 54, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Wurst, W.; Bally-Cuif, L. Neural plate patterning: Upstream and downstream of the isthmic organizer. Nat. Rev. Neurosci. 2001, 2, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.W.; Ross, L.S.; Parrett, T.; Easter, S.S., Jr. The development of a simple scaffold of axon tracts in the brain of the embryonic zebrafish, Brachydanio rerio. Development 1990, 108, 121–145. [Google Scholar] [PubMed]
- Ross, L.S.; Parrett, T.; Easter, S.S., Jr. Axonogenesis and morphogenesis in the embryonic zebrafish brain. J. Neurosci. 1992, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Golden, G.S. A review of the neuroembryology of monoamine systems. Brain Res. Bull. 1982, 9, 553–558. [Google Scholar] [CrossRef]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McLean, D.L.; Fetcho, J.R. Relationship of tyrosine hydroxylase and serotonin immunoreactivity to sensorimotor circuitry in larval zebrafish. J. Comp. Neurol. 2004, 480, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wilson, S.W.; Cooke, S.; Chitnis, A.B.; Driever, W.; Rosenthal, A. Mutations in the zebrafish unmask shared regulatory pathways controlling the development of catecholaminergic neurons. Dev. Biol. 1999, 208, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Holzschuh, J.; Ryu, S.; Aberger, F.; Driever, W. Dopamine transporter expression distinguishes dopaminergic neurons from other catecholaminergic neurons in the developing zebrafish embryo. Mech. Dev. 2001, 101, 237–243. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kani, S.; Shimizu, T.; Tanabe, K.; Nojima, H.; Kimura, Y.; Higashijima, S.; Hibi, M. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev. Biol. 2009, 330, 406–426. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, A.B.; Kuwada, J.Y. Axonogenesis in the brain of zebrafish embryos. J. Neurosci. 1990, 10, 1892–1905. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T. What is the Thalamus in Zebrafish? Front. Neurosci. 2012, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Laessing, U.; Stuermer, C.A. Spatiotemporal pattern of retinal ganglion cell differentiation revealed by the expression of neurolin in embryonic zebrafish. J. Neurobiol. 1996, 29, 65–74. [Google Scholar] [CrossRef]
- Sakata, Y.; Olson, J.K.; Michel, W.C. Assessment of neuronal maturation and acquisition of functional competence in the developing zebrafish olfactory system. Methods Cell Sci. 2003, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Vernier, P.; Wullimann, M.F. A phylotypic stage in vertebrate brain development: GABA cell patterns in zebrafish compared with mouse. J. Comp. Neurol. 2006, 494, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Patterson, J.; Kimmel, R.O. The development and behavioral characteristics of the startle response in the zebra fish. Dev. Psychobiol. 1974, 7, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.; Krumlauf, R. Patterning the vertebrate neuraxis. Science 1996, 274, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Strahle, U.; Scholpp, S. Neurogenesis in zebrafish—From embryo to adult. Neural Dev. 2013, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Strahle, U.; Blader, P. Early neurogenesis in the zebrafish embryo. FASEB J. 1994, 8, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, M.; Bayley, P.; Navratilova, P.; Becker, T.S.; Wilson, S.W.; Clarke, J.D. Morphogenesis underlying the development of the everted teleost telencephalon. Neural Dev. 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Wullimann, M.F. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 2009, 74, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuys, R. The forebrain of actinopterygians revisited. Brain Behav. Evol. 2009, 73, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Ganz, J.; Kroehne, V.; Freudenreich, D.; Machate, A.; Geffarth, M.; Braasch, I.; Kaslin, J.; Brand, M. Subdivisions of the adult zebrafish pallium based on molecular marker analysis. F1000Res 2014, 3, 308. [Google Scholar] [CrossRef] [PubMed]
- Portavella, M.; Torres, B.; Salas, C.; Papini, M.R. Lesions of the medial pallium, but not of the lateral pallium, disrupt spaced-trial avoidance learning in goldfish (Carassius auratus). Neurosci. Lett. 2004, 362, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.; Tanabe, H.; Suster, M.L.; Ailani, D.; Kotani, Y.; Muto, A.; Itoh, M.; Iwasaki, M.; Wada, H.; Yaksi, E.; et al. Identification of a neuronal population in the telencephalon essential for fear conditioning in zebrafish. BMC Biol. 2018, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Broglio, C.; Gomez, A.; Duran, E.; Ocana, F.M.; Jimenez-Moya, F.; Rodriguez, F.; Salas, C. Hallmarks of a common forebrain vertebrate plan: Specialized pallial areas for spatial, temporal and emotional memory in actinopterygian fish. Brain Res. Bull. 2005, 66, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Broglio, C.; Rodriguez, F.; Gomez, A.; Arias, J.L.; Salas, C. Selective involvement of the goldfish lateral pallium in spatial memory. Behav. Brain Res. 2010, 210, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Ullmann, J.F.; Norton, W.H.; Parker, M.O.; Brennan, C.H.; Gerlai, R.; Kalueff, A.V. Molecular psychiatry of zebrafish. Mol. Psychiatry 2015, 20, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.H. Toward developmental models of psychiatric disorders in zebrafish. Front. Neural Circuits 2013, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Wullimann, M.F.; Mueller, T.; Distel, M.; Babaryka, A.; Grothe, B.; Koster, R.W. The long adventurous journey of rhombic lip cells in jawed vertebrates: A comparative developmental analysis. Front. Neuroanat 2011, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamaguchi, S.; Sakakibara, Y.; Hayashi, T.; Matsuda, K.; Hara, Y.; Tanegashima, C.; Shimizu, T.; Kuraku, S.; Hibi, M. Gene expression profiling of granule cells and Purkinje cells in the zebrafish cerebellum. J. Comp. Neurol. 2017, 525, 1558–1585. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Namikawa, K.; Babaryka, A.; Koster, R.W. Functional regionalization of the teleost cerebellum analyzed in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 11846–11851. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, R.V.; Joyner, A.L. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu. Rev. Cell Dev. Biol. 2007, 23, 549–577. [Google Scholar] [CrossRef] [PubMed]
- Kani, S.; Bae, Y.K.; Shimizu, T.; Tanabe, K.; Satou, C.; Parsons, M.J.; Scott, E.; Higashijima, S.; Hibi, M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010, 343, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hibi, M.; Matsuda, K.; Takeuchi, M.; Shimizu, T.; Murakami, Y. Evolutionary mechanisms that generate morphology and neural-circuit diversity of the cerebellum. Dev. Growth Differ. 2017, 59, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, S.J.G.; Lee, M.M.; Ramirez, A.D.; Lim, S.; Goldman, M.S.; Aksay, E.R.F. Population-scale organization of cerebellar granule neuron signaling during a visuomotor behavior. Sci. Rep. 2017, 7, 16240. [Google Scholar] [CrossRef] [PubMed]
- Severi, K.E.; Bohm, U.L.; Wyart, C. Investigation of hindbrain activity during active locomotion reveals inhibitory neurons involved in sensorimotor processing. Sci. Rep. 2018, 8, 13615. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, A.; Zee, D.S. Cerebellum and ocular motor control. Front. Neurol. 2011, 2, 53. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.M.; Cook, E.H.; Sweeney, J.A.; Mosconi, M.W. Saccadic eye movement abnormalities in autism spectrum disorder indicate dysfunctions in cerebellum and brainstem. Mol. Autism 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.Y.; Ulrich, B.; Issa, F.A.; Wan, J.; Papazian, D.M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Front. Neural Circuits 2014, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.E.; Turner, R.W. Physiological and morphological development of the rat cerebellar Purkinje cell. J. Physiol. 2005, 567, 829–850. [Google Scholar] [CrossRef] [PubMed]
- Tropepe, V.; Sive, H.L. Can zebrafish be used as a model to study the neurodevelopmental causes of autism? Genes Brain Behav. 2003, 2, 268–281. [Google Scholar] [CrossRef] [PubMed]
- De Rienzo, G.; Bishop, J.A.; Mao, Y.; Pan, L.; Ma, T.P.; Moens, C.B.; Tsai, L.H.; Sive, H. Disc1 regulates both beta-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J. 2011, 25, 4184–4197. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, S.; Hoffman, E.J. Zebrafish: A Translational Model System for Studying Neuropsychiatric Disorders. J. Am. Acad. Child. Adolesc. Psychiatry 2016, 55, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef] [PubMed]
- Blaker-Lee, A.; Gupta, S.; McCammon, J.M.; De Rienzo, G.; Sive, H. Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model Mech. 2012, 5, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Elsen, G.E.; Choi, L.Y.; Prince, V.E.; Ho, R.K. The autism susceptibility gene met regulates zebrafish cerebellar development and facial motor neuron migration. Dev. Biol. 2009, 335, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Psychiatric, G.C.C.C.; Cichon, S.; Craddock, N.; Daly, M.; Faraone, S.V.; Gejman, P.V.; Kelsoe, J.; Lehner, T.; Levinson, D.F.; Moran, A.; et al. Genomewide association studies: History, rationale, and prospects for psychiatric disorders. Am. J. Psychiatry 2009, 166, 540–556. [Google Scholar]
- Volkmar, F.R.; McPartland, J.C. From Kanner to DSM-5: Autism as an evolving diagnostic concept. Annu. Rev. Clin. Psychol. 2014, 10, 193–212. [Google Scholar] [CrossRef] [PubMed]
- DiBella, L.M.; Park, A.; Sun, Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 2009, 18, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Richards, R.I.; Lardelli, M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 2006, 15, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Champagne, N.; Lafreniere, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Cote, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Holper, S.; Kruger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Spitzer, N.C. Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 2010, 33, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar] [PubMed]
- Lejeune, J.; Maunoury, C.; Prieur, M.; Van den Akker, J. A jumping translocation (5p;15q), (8q;15q), and (12q;15q) (author’s transl). Ann. Genet. 1979, 22, 210–213. [Google Scholar] [PubMed]
- Archidiacono, N.; Rett, A.; Rocchi, M.; Rolando, S.; Lugaresi, E.; Romeo, G. Rett syndrome and fragile site in Xp22. Lancet 1985, 2, 1242–1243. [Google Scholar] [CrossRef]
- Fryer, A.E.; Chalmers, A.; Connor, J.M.; Fraser, I.; Povey, S.; Yates, A.D.; Yates, J.R.; Osborne, J.P. Evidence that the gene for tuberous sclerosis is on chromosome 9. Lancet 1987, 1, 659–661. [Google Scholar] [CrossRef]
- Kandt, R.S.; Haines, J.L.; Smith, M.; Northrup, H.; Gardner, R.J.; Short, M.P.; Dumars, K.; Roach, E.S.; Steingold, S.; Wall, S.; et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat. Genet. 1992, 2, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.B.; Konecki, D.S.; Munstermann, E.; Bolton, P.; Poustka, A.; Poustka, F.; Gurling, H.M. Point mutation analysis of the FMR-1 gene in autism. Mol. Psychiatry 1996, 1, 227–231. [Google Scholar] [PubMed]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; D’Agruma, L.; Maiorano, N.; Totaro, A.; Militerni, R.; Bravaccio, C.; Wassink, T.H.; Schneider, C.; Melmed, R.; Trillo, S.; et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 2001, 6, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Escayg, A.; Kearney, J.A.; Trudeau, M.; MacDonald, B.T.; Mori, M.; Reichert, J.; Buxbaum, J.D.; Meisler, M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 2003, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Giorda, R.; Mani, E.; Aceti, G.; Anderlid, B.M.; Baroncini, A.; Pramparo, T.; Zuffardi, O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006, 43, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Vrijenhoek, T.; Buizer-Voskamp, J.E.; van der Stelt, I.; Strengman, E.; Genetic, R.; Outcome in Psychosis, C.; Sabatti, C.; Geurts van Kessel, A.; Brunner, H.G.; Ophoff, R.A.; et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 2008, 83, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Nord, A.S.; Roeb, W.; Dickel, D.E.; Walsh, T.; Kusenda, M.; O’Connor, K.L.; Malhotra, D.; McCarthy, S.E.; Stray, S.M.; Taylor, S.M.; et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 2011, 19, 727–731. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.H.; Yu, T.W.; Lim, E.T.; Ataman, B.; Coulter, M.E.; Hill, R.S.; Stevens, C.R.; Schubert, C.R.; Collaboration, A.A.S.; Greenberg, M.E.; et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012, 8, e1002635. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, D.L.; Soden, S.E.; Saunders, C.J.; Miller, N.A.; Farrow, E.G.; Smith, L.D.; Kingsmore, S.F. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med. Genom. 2013, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Suls, A.; Jaehn, J.A.; Kecskes, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.C.; Siekierska, A.; Djemie, T.; Afrikanova, T.; Gormley, P.; et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, W.; Nilsson, D.; Topa, A.; Anderlid, B.M.; Darki, F.; Matsson, H.; Tapia Paez, I.; Klingberg, T.; Samuelsson, L.; Wirta, V.; et al. CTNND2-a candidate gene for reading problems and mild intellectual disability. J. Med. Genet. 2015, 52, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Warga, R.M.; Kimmel, C.B. Cell movements during epiboly and gastrulation in zebrafish. Development 1990, 108, 569–580. [Google Scholar] [PubMed]
- Ho, R.K.; Kimmel, C.B. Commitment of cell fate in the early zebrafish embryo. Science 1993, 261, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.; Gates, M.A.; Egan, E.S.; Dougan, S.T.; Rennebeck, G.; Sirotkin, H.I.; Schier, A.F.; Talbot, W.S. Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature 1998, 395, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, D.; Klingensmith, J.; Kemp, C.; Belo, J.A.; Anderson, R.M.; May, S.R.; McMahon, J.A.; McMahon, A.P.; Harland, R.M.; Rossant, J.; et al. The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature 2000, 403, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Houart, C.; Westerfield, M.; Wilson, S.W. A small population of anterior cells patterns the forebrain during zebrafish gastrulation. Nature 1998, 391, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Lun, K.; Brand, M. A series of no isthmus (noi) alleles of the zebrafish pax2.1 gene reveals multiple signaling events in development of the midbrain-hindbrain boundary. Development 1998, 125, 3049–3062. [Google Scholar] [PubMed]
- Grinblat, Y.; Gamse, J.; Patel, M.; Sive, H. Determination of the zebrafish forebrain: Induction and patterning. Development 1998, 125, 4403–4416. [Google Scholar] [PubMed]
- Brand, M.; Heisenberg, C.P.; Jiang, Y.J.; Beuchle, D.; Lun, K.; Furutani-Seiki, M.; Granato, M.; Haffter, P.; Hammerschmidt, M.; Kane, D.A.; et al. Mutations in zebrafish genes affecting the formation of the boundary between midbrain and hindbrain. Development 1996, 123, 179–190. [Google Scholar] [PubMed]
- Piccolo, S.; Sasai, Y.; Lu, B.; De Robertis, E.M. Dorsoventral patterning in Xenopus: Inhibition of ventral signals by direct binding of chordin to BMP-4. Cell 1996, 86, 589–598. [Google Scholar] [CrossRef]
- Turner, T.N.; Sharma, K.; Oh, E.C.; Liu, Y.P.; Collins, R.L.; Sosa, M.X.; Auer, D.R.; Brand, H.; Sanders, S.J.; Moreno-De-Luca, D.; et al. Loss of delta-catenin function in severe autism. Nature 2015, 520, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Sokol, S.Y. Graded amounts of Xenopus dishevelled specify discrete anteroposterior cell fates in prospective ectoderm. Mech. Dev. 1997, 61, 113–125. [Google Scholar] [CrossRef]
- Backman, M.; Machon, O.; Mygland, L.; van den Bout, C.J.; Zhong, W.; Taketo, M.M.; Krauss, S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev. Biol. 2005, 279, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–4477. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, A.; Nowosiad, P.; Jagasia, R.; Aigner, S.; Taylor, R.D.; Andreae, L.C.; Gatford, N.J.F.; Lucchesi, W.; Srivastava, D.P.; Price, J. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 2018, 23, 735–746. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.M.; Blaker-Lee, A.; Chen, X.; Sive, H. The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet. 2017, 26, 3699–3712. [Google Scholar] [CrossRef] [PubMed]
- Escamilla, C.O.; Filonova, I.; Walker, A.K.; Xuan, Z.X.; Holehonnur, R.; Espinosa, F.; Liu, S.; Thyme, S.B.; Lopez-Garcia, I.A.; Mendoza, D.B.; et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 2017, 551, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, J.Y.; Bernhardt, R.R. Axonal outgrowth by identified neurons in the spinal cord of zebrafish embryos. Exp. Neurol. 1990, 109, 29–34. [Google Scholar] [CrossRef]
- Metcalfe, W.K.; Myers, P.Z.; Trevarrow, B.; Bass, M.B.; Kimmel, C.B. Primary neurons that express the L2/HNK-1 carbohydrate during early development in the zebrafish. Development 1990, 110, 491–504. [Google Scholar] [PubMed]
- Borodinsky, L.N.; Root, C.M.; Cronin, J.A.; Sann, S.B.; Gu, H.L.; Spitzer, N.C. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 2004, 429, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, N.C. Electrical activity in early neuronal development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Gatto, C.L.; Broadie, K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2010, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.F.; Yang, L.; Li, S.; Wang, Q.; Yuan, X.B.; Gao, X.; Bao, L.; Zhang, X. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 2012, 149, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; De Rienzo, G.; Drane, L.; Mao, Y.; Flood, Z.; Madison, J.; Ferreira, M.; Bergen, S.; King, C.; Sklar, P.; et al. Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 2011, 72, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Golden, C.E.; Buxbaum, J.D.; De Rubeis, S. Disrupted circuits in mouse models of autism spectrum disorder and intellectual disability. Curr. Opin. Neurobiol. 2018, 48, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Dutton, S.B.; Makinson, C.D.; Papale, L.A.; Shankar, A.; Balakrishnan, B.; Nakazawa, K.; Escayg, A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 2013, 49, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- Herget, U.; Gutierrez-Triana, J.A.; Salazar Thula, O.; Knerr, B.; Ryu, S. Single-Cell Reconstruction of Oxytocinergic Neurons Reveals Separate Hypophysiotropic and Encephalotropic Subtypes in Larval Zebrafish. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Herget, U.; Wolf, A.; Wullimann, M.F.; Ryu, S. Molecular neuroanatomy and chemoarchitecture of the neurosecretory preoptic-hypothalamic area in zebrafish larvae. J. Comp. Neurol. 2014, 522, 1542–1564. [Google Scholar] [CrossRef] [PubMed]
- Filippi, A.; Mueller, T.; Driever, W. vglut2 and gad expression reveal distinct patterns of dual GABAergic versus glutamatergic cotransmitter phenotypes of dopaminergic and noradrenergic neurons in the zebrafish brain. J. Comp. Neurol. 2014, 522, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Haehnel-Taguchi, M.; Fernandes, A.M.; Bohler, M.; Schmitt, I.; Tittel, L.; Driever, W. Projections of the Diencephalospinal Dopaminergic System to Peripheral Sense Organs in Larval Zebrafish (Danio rerio). Front. Neuroanat. 2018, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.; Kontarakis, Z.; Rossi, A. Making sense of anti-sense data. Dev. Cell 2015, 32, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Postlethwait, J.; Amores, A.; Cresko, W.; Singer, A.; Yan, Y.L. Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. 2004, 20, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.M.; Braasch, I.; Postlethwait, J.H. Conserved synteny and the zebrafish genome. Methods Cell. Biol. 2011, 104, 259–285. [Google Scholar] [PubMed]
- Pasquier, J.; Braasch, I.; Batzel, P.; Cabau, C.; Montfort, J.; Nguyen, T.; Jouanno, E.; Berthelot, C.; Klopp, C.; Journot, L.; et al. Evolution of gene expression after whole-genome duplication: New insights from the spotted gar genome. J. Exp. Zool. B Mol. Dev. Evol. 2017, 328, 709–721. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, M.L.; Monani, U.R.; Burghes, A.H.; Beattie, C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003, 162, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Shepard, J.L.; Amatruda, J.F.; Stern, H.M.; Subramanian, A.; Finkelstein, D.; Ziai, J.; Finley, K.R.; Pfaff, K.L.; Hersey, C.; Zhou, Y.; et al. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc. Natl. Acad. Sci. USA 2005, 102, 13194–13199. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Einhorn, Z.; Mercurio, S.; Lee, S.; Lau, B.; Mione, M.; Wilson, S.W.; Guo, S. Neurogenin1 is a determinant of zebrafish basal forebrain dopaminergic neurons and is regulated by the conserved zinc finger protein Tof/Fezl. Proc. Natl. Acad. Sci. USA 2006, 103, 5143–5148. [Google Scholar] [CrossRef] [PubMed]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, e78. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Karlen, S.; Rebagliati, M. A morpholino phenocopy of the cyclops mutation. Genesis 2001, 30, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, L.; Page-McCaw, P.S.; Chen, W. Zebrafish Genome Engineering Using the CRISPR-Cas9 System. Trends Genet. 2016, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Simone, B.W.; Martinez-Galvez, G.; WareJoncas, Z.; Ekker, S.C. Fishing for understanding: Unlocking the zebrafish gene editor’s toolbox. Methods 2018, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.S.; Dahlem, T.J.; Bisgrove, B.W.; Yost, H.J.; Tristani-Firouzi, M. CRISPR/Cas9-Directed Gene Editing for the Generation of Loss-of-Function Mutants in High-Throughput Zebrafish F0 Screens. Curr. Protoc. Mol. Biol. 2017, 119, 31–39. [Google Scholar] [PubMed]
- Shah, A.N.; Moens, C.B.; Miller, A.C. Targeted candidate gene screens using CRISPR/Cas9 technology. Methods Cell Biol. 2016, 135, 89–106. [Google Scholar] [PubMed]
- Saint-Amant, L.; Drapeau, P. Time course of the development of motor behaviors in the zebrafish embryo. J. Neurobiol. 1998, 37, 622–632. [Google Scholar] [CrossRef]
- Grillner, S. The motor infrastructure: From ion channels to neuronal networks. Nat. Rev. Neurosci. 2003, 4, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Goulding, M.D. Circuits controlling vertebrate locomotion: Moving in a new direction. Nat. Rev. Neurosci. 2009, 10, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.A.; Granato, M. Sensorimotor gating in larval zebrafish. J. Neurosci. 2007, 27, 4984–4994. [Google Scholar] [CrossRef] [PubMed]
- Portugues, R.; Feierstein, C.E.; Engert, F.; Orger, M.B. Whole-brain activity maps reveal stereotyped, distributed networks for visuomotor behavior. Neuron 2014, 81, 1328–1343. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, T.; Bellesheim, K.; Siper, P.M.; Wang, A.T.; Halpern, D.; Gorenstein, M.; Grodberg, D.; Kolevzon, A.; Buxbaum, J.D. Measuring Sensory Reactivity in Autism Spectrum Disorder: Application and Simplification of a Clinician-Administered Sensory Observation Scale. J. Autism. Dev. Disord. 2016, 46, 287–293. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association; American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed. American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Engeszer, R.E.; Barbiano, L.A.; Ryan, M.J.; Parichy, D.M. Timing and plasticity of shoaling behaviour in the zebrafish, Danio rerio. Anim. Behav. 2007, 74, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Dreosti, E.; Lopes, G.; Kampff, A.R.; Wilson, S.W. Development of social behavior in young zebrafish. Front. Neural Circuits 2015, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, G.; Hodgins-Davis, A.; Avolio, C.; Schunter, C. Kin recognition in zebrafish: A 24-hour window for olfactory imprinting. Proc. Biol. Sci. 2008, 275, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Wircer, E.; Blechman, J.; Borodovsky, N.; Tsoory, M.; Nunes, A.R.; Oliveira, R.F.; Levkowitz, G. Homeodomain protein Otp affects developmental neuropeptide switching in oxytocin neurons associated with a long-term effect on social behavior. Elife 2017, 6, e22170. [Google Scholar] [CrossRef] [PubMed]
- Engeszer, R.E.; Ryan, M.J.; Parichy, D.M. Learned social preference in zebrafish. Curr. Biol. 2004, 14, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Stednitz, S.J.; McDermott, E.M.; Ncube, D.; Tallafuss, A.; Eisen, J.S.; Washbourne, P. Forebrain Control of Behaviorally Driven Social Orienting in Zebrafish. Curr. Biol. 2018, 28, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.; Anderlid, B.M.; Nordgren, A.; Willfors, C.; Kuja-Halkola, R.; Tammimies, K.; Bolte, S. Minor physical anomalies in neurodevelopmental disorders: A twin study. Child. Adolesc. Psychiatry Ment. Health 2017, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, R.; DeVito, T.J.; Vidal, C.N.; Sui, Y.; Hayashi, K.M.; Drost, D.J.; Williamson, P.C.; Rajakumar, N.; Toga, A.W.; Thompson, P.M. Detection and mapping of hippocampal abnormalities in autism. Psychiatry Res. 2006, 148, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Randlett, O.; Wee, C.L.; Naumann, E.A.; Nnaemeka, O.; Schoppik, D.; Fitzgerald, J.E.; Portugues, R.; Lacoste, A.M.; Riegler, C.; Engert, F.; et al. Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 2015, 12, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Marquart, G.D.; Horstick, E.J.; Tabor, K.M.; Pajevic, S.; Burgess, H.A. Morphometric analysis and neuroanatomical mapping of the zebrafish brain. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Marquart, G.D.; Tabor, K.M.; Horstick, E.J.; Brown, M.; Geoca, A.K.; Polys, N.F.; Nogare, D.D.; Burgess, H.A. High-precision registration between zebrafish brain atlases using symmetric diffeomorphic normalization. Gigascience 2017, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Forster, D.; Arnold-Ammer, I.; Laurell, E.; Barker, A.J.; Fernandes, A.M.; Finger-Baier, K.; Filosa, A.; Helmbrecht, T.O.; Kolsch, Y.; Kuhn, E.; et al. Genetic targeting and anatomical registration of neuronal populations in the zebrafish brain with a new set of BAC transgenic tools. Sci. Rep. 2017, 7, 5230. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, G.S.; Potter, C.J.; Chan, A.M.; Marin, E.C.; Rohlfing, T.; Maurer, C.R., Jr.; Luo, L. Comprehensive maps of Drosophila higher olfactory centers: Spatially segregated fruit and pheromone representation. Cell 2007, 128, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Satou, C.; Kimura, Y.; Higashijima, S. Generation of multiple classes of V0 neurons in zebrafish spinal cord: Progenitor heterogeneity and temporal control of neuronal diversity. J. Neurosci. 2012, 32, 1771–1783. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.F.; Timlin, J.A.; Roysam, B.; McNaughton, B.L.; Worley, P.F.; Barnes, C.A. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr. Opin. Neurobiol. 2005, 15, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Barros, V.N.; Mundim, M.; Galindo, L.T.; Bittencourt, S.; Porcionatto, M.; Mello, L.E. The pattern of c-Fos expression and its refractory period in the brain of rats and monkeys. Front. Cell Neurosci. 2015, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dudek, H.; Miranti, C.K.; Greenberg, M.E. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996, 16, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.J.; Fetcho, J.R. Labeling blastomeres with a calcium indicator: A non-invasive method of visualizing neuronal activity in zebrafish. J. Neurosci. Methods 1996, 68, 185–191. [Google Scholar] [CrossRef]
- Takahashi, M.; Narushima, M.; Oda, Y. In vivo imaging of functional inhibitory networks on the mauthner cell of larval zebrafish. J. Neurosci. 2002, 22, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Sumbre, G.; Muto, A.; Baier, H.; Poo, M.M. Entrained rhythmic activities of neuronal ensembles as perceptual memory of time interval. Nature 2008, 456, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, J.; Marques, J.C.; Grama, A.; Hildebrand, D.G.C.; Gu, W.; Li, J.M.; Robson, D.N. Pan-neuronal calcium imaging with cellular resolution in freely swimming zebrafish. Nat. Methods 2017, 14, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Wang, Z.; Chai, Y.; Hang, W.; Shang, C.; Yang, W.; Bai, L.; Du, J.; Wang, K.; Wen, Q. Rapid whole brain imaging of neural activity in freely behaving larval zebrafish (Danio rerio). Elife 2017, 6, e28158. [Google Scholar] [CrossRef] [PubMed]
- Lovett-Barron, M.; Andalman, A.S.; Allen, W.E.; Vesuna, S.; Kauvar, I.; Burns, V.M.; Deisseroth, K. Ancestral Circuits for the Coordinated Modulation of Brain State. Cell 2017, 171, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Jawinski, P.; Kirsten, H.; Sander, C.; Spada, J.; Ulke, C.; Huang, J.; Burkhardt, R.; Scholz, M.; Hensch, T.; Hegerl, U. Human brain arousal in the resting state: A genome-wide association study. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Mollinedo-Gajate, I.; Penagarikano, O. Neural Circuits for Social Cognition: Implications for Autism. Neuroscience 2018, 370, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Asakawa, K.; Hibi, M.; Itoh, M.; Muto, A.; Wada, H. Gal4 Driver Transgenic Zebrafish: Powerful Tools to Study Developmental Biology, Organogenesis, and Neuroscience. Adv. Genet. 2016, 95, 65–87. [Google Scholar] [PubMed]
- Wiley, D.S.; Redfield, S.E.; Zon, L.I. Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell Biol. 2017, 138, 651–679. [Google Scholar] [PubMed]
- Svenson, K.L.; Gatti, D.M.; Valdar, W.; Welsh, C.E.; Cheng, R.; Chesler, E.J.; Palmer, A.A.; McMillan, L.; Churchill, G.A. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics 2012, 190, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Rihel, J.; Prober, D.A.; Arvanites, A.; Lam, K.; Zimmerman, S.; Jang, S.; Haggarty, S.J.; Kokel, D.; Rubin, L.L.; Peterson, R.T.; et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Kokel, D.; Peterson, R.T. Using the zebrafish photomotor response for psychotropic drug screening. Methods Cell. Biol. 2011, 105, 517–524. [Google Scholar] [PubMed]
- Bruni, G.; Rennekamp, A.J.; Velenich, A.; McCarroll, M.; Gendelev, L.; Fertsch, E.; Taylor, J.; Lakhani, P.; Lensen, D.; Evron, T.; et al. Zebrafish behavioral profiling identifies multitarget antipsychotic-like compounds. Nat. Chem. Biol. 2016, 12, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.; Beresford, C.A.; Bunker, M.; Verdi, M.; Vishnevetsky, D.; Karlsson, C.; Teer, O.; Stedman, A.; Smith, K.A. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J. Child Adolesc. Psychopharmacol. 2014, 24, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.E.; Gregas, M.; Jeste, S.S. Risperidone use in autism spectrum disorders: A retrospective review of a clinic-referred patient population. J. Child Neurol. 2011, 26, 428–432. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Structure or Region | Estimated Onset of Development (dpf) | ||
|---|---|---|---|
| Human [75,78] | Rat [75,78] | Zebrafish | |
| Neural tube | 21 | 10 | 0.4–0.8 [14] |
| Telencephalon, Diencephalon, Mesencephalon, Rhombencephalon | 28–33 | 11 | 0.7–0.75 [14] |
| Cerebellar primordia | 28 | 12 | 0.8 [14] |
| Optic tectum | 28 | 12 | 1 [82] |
| Hypothalamus, pineal (epiphysis) | 28 | 12 | 0.75, 1 [83] |
| Raphe complex | 28 [84] | 10.5 [85] | 3 [86] |
| Locus coeruleus | 28 | 11 | 1 [87,88] |
| Inferior olive | 30 | 11 | 3 [89] |
| Medial longitudinal fasciculus | 30 | 11 | 1.2 [90] |
| Thalamus | 36 | 14 | 2 [91] |
| Cell type | |||
| Retinal ganglion cells | 33 | 12 | 1.2 [92] |
| Mitral cells | 33 | 12 | 1 [93] |
| GABAergic neurons | 52 | 15 | 1.2 [94] |
| Behavior | |||
| Walking (zebrafish, swimming) | 455 | 48 | 1.13 [23] |
| Acoustic startle response | 265 | 29 | 4 [95] |
| Developmental Event | Estimated Onset of Development (dpf) | |
|---|---|---|
| Rat [112] | Zebrafish [109,113,119] | |
| Cerebellar primordia (En1/2, Wnt1, Pax2, Gbx2) | 12.5 | 0.8 |
| Progenitor pools specified (Atoh1a/Ptf1a) | 10.5 | 1–2 |
| GC/PC migration-differentiation | 12.5–15 | 3 |
| Purkinje cells functionally mature | 29.5 [120] | 4 |
| Trilaminar structure formed | 35–42 | 5 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozol, R.A. Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model. J. Dev. Biol. 2018, 6, 29. https://doi.org/10.3390/jdb6040029
Kozol RA. Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model. Journal of Developmental Biology. 2018; 6(4):29. https://doi.org/10.3390/jdb6040029
Chicago/Turabian StyleKozol, Robert A. 2018. "Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model" Journal of Developmental Biology 6, no. 4: 29. https://doi.org/10.3390/jdb6040029
APA StyleKozol, R. A. (2018). Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model. Journal of Developmental Biology, 6(4), 29. https://doi.org/10.3390/jdb6040029