Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
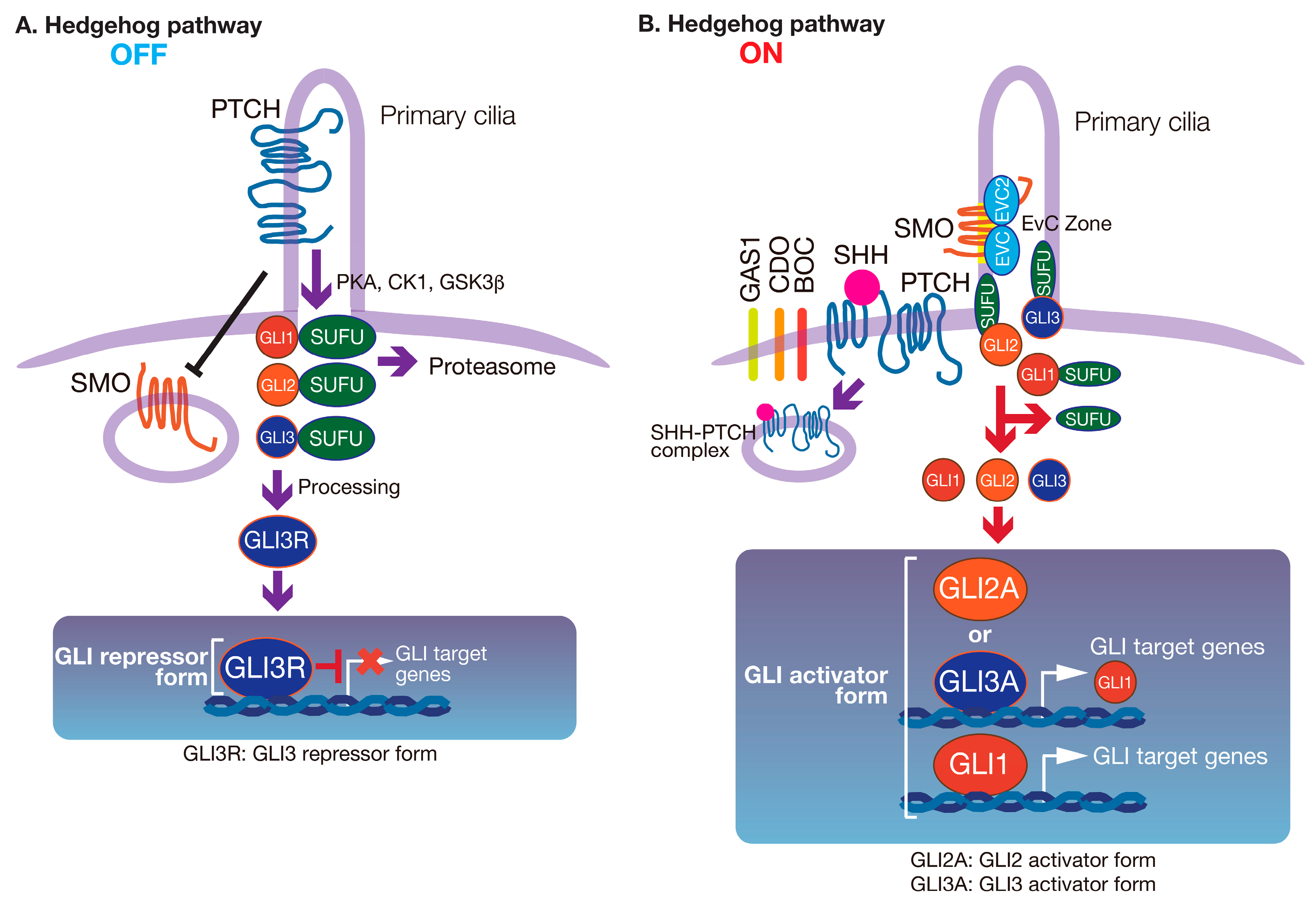
2. The HH Signaling Pathway
3. Overview of Epidermal and Hair Follicle Development
3.1. Development of the Epidermis
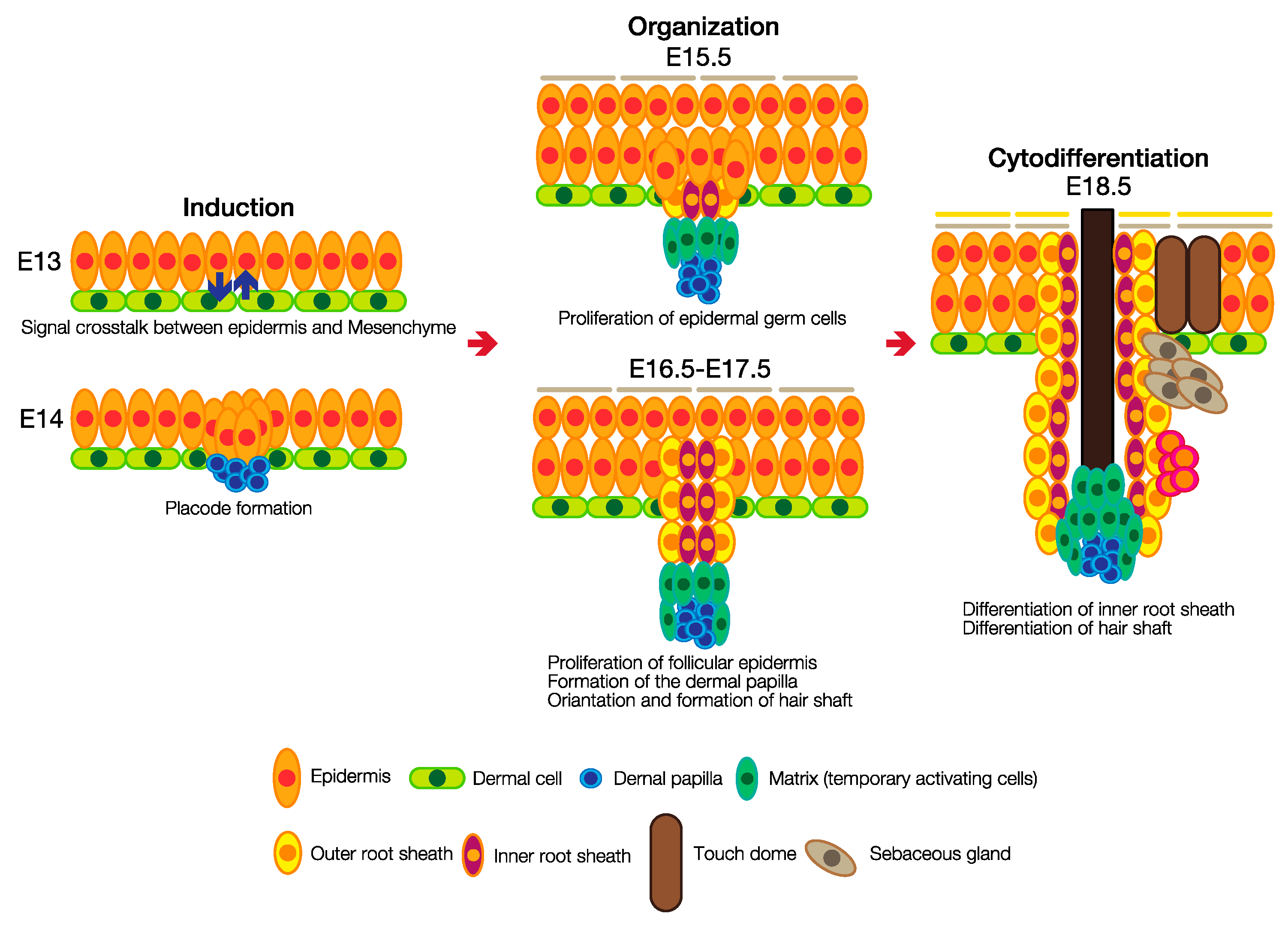
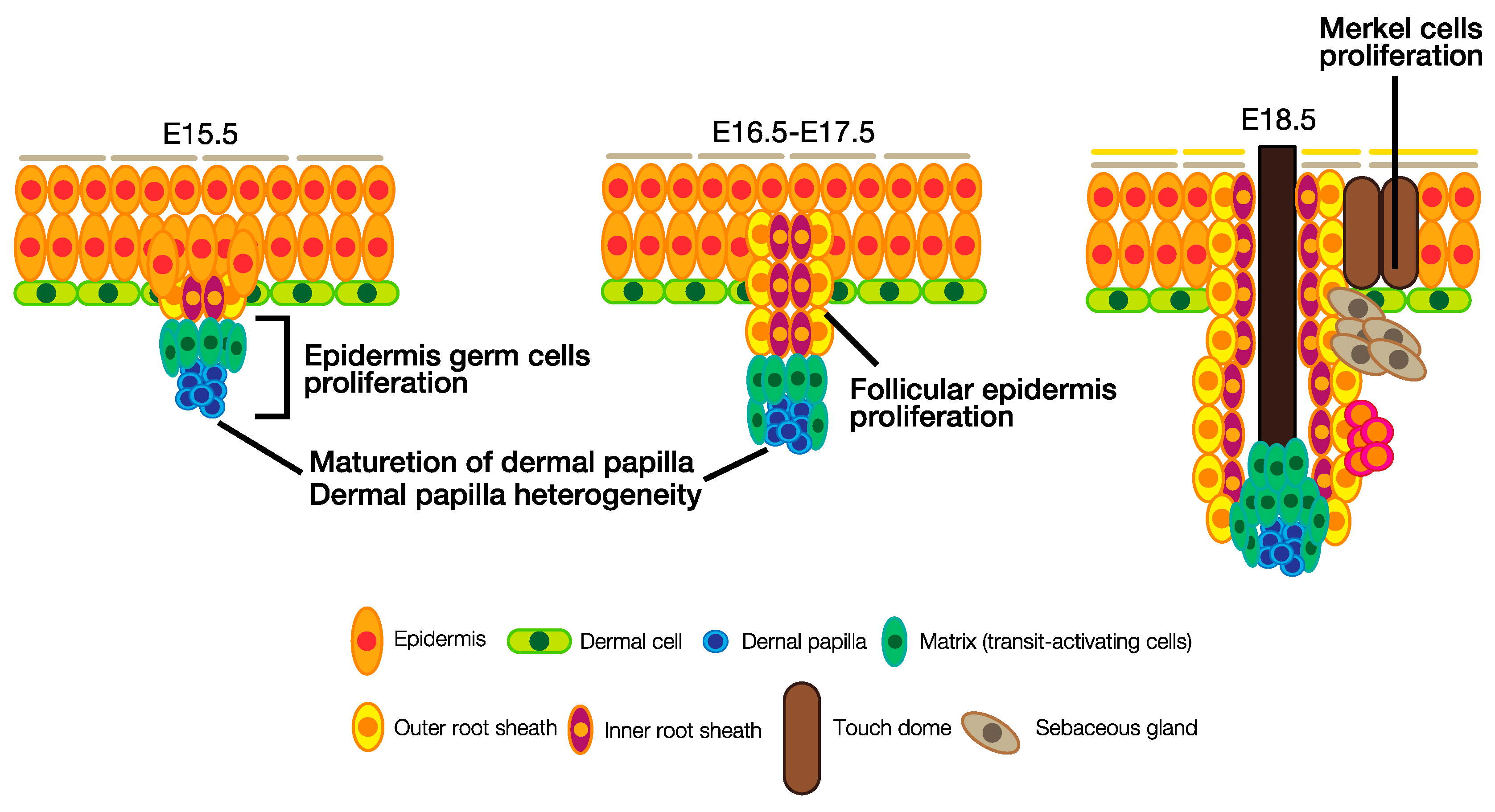
3.2. Hair Follicle Development
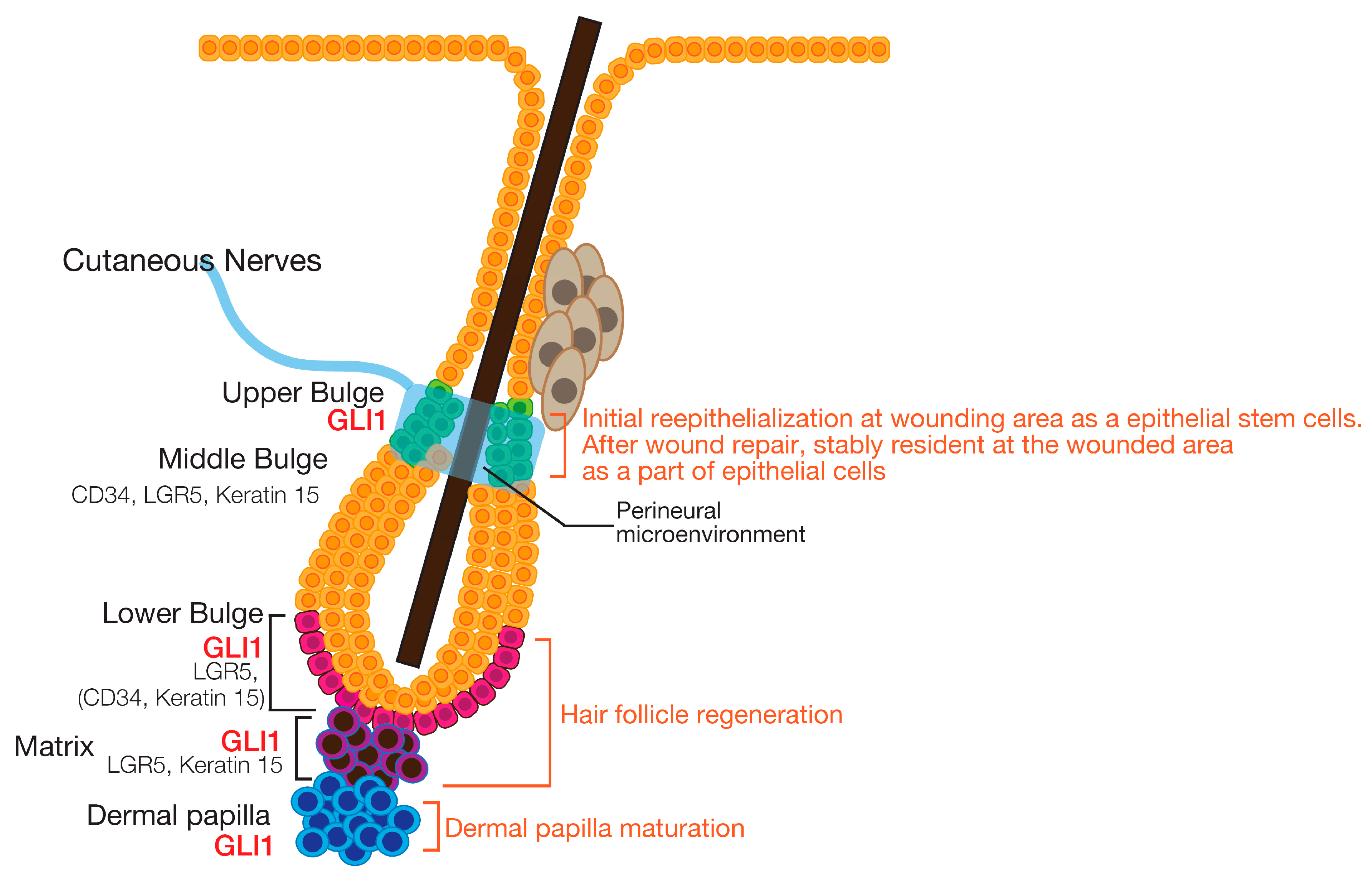
4. The Roles of HH Signaling in Hair Follicle and Touch Dome Development
5. Signaling Crosstalk between HH and Other Pathways in Hair Follicle Development
5.1. Wnt Signaling
5.2. BMP Signaling
5.3. PDGF Signaling
5.4. Notch Signaling
5.5. The EdaA1–EdaR–NF-κB Axis
6. Epidermal Homeostasis
7. Roles of the HH Signaling Pathway in Epidermal Homeostasis
8. Signaling Crosstalk between HH and Other Signaling Pathways in Epidermal Homeostasis
8.1. Wnt Signaling
8.2. The Hippo–YAP Signaling Pathway
8.3. p53/p63
9. Aberrant HH Signaling Pathway Activation and BCC
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koster, M.I.; Roop, D.R. Mechanisms Regulating Epithelial Stratification. Annu. Rev. Cell Dev. Biol. 2007, 23, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ullrich, R.; Paus, R. Molecular principles of hair follicle induction and morphogenesis. BioEssays 2005, 27, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Dlugosz, A.; Agrawal, S.; Kirkpatrick, P. Vismodegib. Nat. Rev. Drug Discov. 2012, 11, 437–438. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; i Altaba, A.R. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Charron, F.; Morin, S.; Shin, D.S.; Wong, K.; Fabre, P.J.; Tessier-Lavigne, M.; McConnell, S.K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 2006, 444, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 Complex Transduces the Hedgehog Signal at Primary Cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. BioMed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development (Camb. Engl.) 1999, 126, 3915–3924. [Google Scholar]
- Bai, C.B.; Auerbach, W.; Lee, J.S.; Stephen, D.; Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development (Camb. Engl.) 2002, 129, 4753–4761. [Google Scholar]
- Masuya, H.; Sagai, T.; Moriwaki, K.; Shiroishi, T. Multigenic control of the localization of the zone of polarizing activity in limb morphogenesis in the mouse. Dev. Biol. 1997, 182, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Büscher, D.; Bosse, B.; Heymer, J.; Rüther, U. Evidence for genetic control of Sonic hedgehog by Gli3 in mouse limb development. Mech. Dev. 1997, 62, 175–182. [Google Scholar] [CrossRef]
- Ding, Q.; Motoyama, J.; Gasca, S.; Mo, R.; Sasaki, H.; Rossant, J.; Hui, C.C. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development (Camb. Engl.) 1998, 125, 2533–2543. [Google Scholar]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Park, H.L.; Bai, C.; Platt, K.A.; Matise, M.P.; Beeghly, A.; Hui, C.C.; Nakashima, M.; Joyner, A.L. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development (Camb. Engl.) 2000, 127, 1593–1605. [Google Scholar] [CrossRef]
- Bai, C.B.; Joyner, A.L. Gli1 can rescue the in vivo function of Gli2. Development (Camb. Engl.) 2001, 128, 5161–5172. [Google Scholar]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Wolter, M.; Reifenberger, J.; Sommer, C.; Ruzicka, T.; Reifenberger, G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997, 57, 2581–2585. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef] [PubMed]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. β-Catenin Controls Hair Follicle Morphogenesis and Stem Cell Differentiation in the Skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamaguchi, Y.; Villacorte, M.; Mihara, K.; Akiyama, M.; Shimizu, H.; Taketo, M.M.; Nakagata, N.; Tsukiyama, T.; Yamaguchi, T.P.; et al. Embryonic hair follicle fate change by augmented -catenin through Shh and Bmp signaling. Development 2009, 136, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Gat, U.; DasGupta, R.; Degenstein, L.; Fuchs, E. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 1998, 95, 605–614. [Google Scholar] [CrossRef]
- Silva-Vargas, V.; Lo Celso, C.; Giangreco, A.; Ofstad, T.; Prowse, D.M.; Braun, K.M.; Watt, F.M. β-Catenin and Hedgehog Signal Strength Can Specify Number and Location of Hair Follicles in Adult Epidermis without Recruitment of Bulge Stem Cells. Dev. Cell 2005, 9, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Pierrat, M.-J.; Mauviel, A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.-C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.-K.; Kittappa, R.; McKay, R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, M.L. Genetic basis of skin appendage development. Semin. Cell Dev. Biol. 2007, 18, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, J.; Takabatake, T.; Takeshima, K.; Hui, C. Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 1998, 18, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Müller-Röver, S.; Van Der Veen, C.; Maurer, M.; Eichmüller, S.; Ling, G.; Hofmann, U.; Foitzik, K.; Mecklenburg, L.; Handjiski, B. A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J. Investig. Dermatol. 1999, 113, 523–532. [Google Scholar] [PubMed]
- Forni, M.F.; Trombetta-Lima, M.; Sogayar, M.C. Stem cells in embryonic skin development. Biol. Res. 2012, 45, 215–222. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 1998, 8, 1058–1068. [Google Scholar] [CrossRef]
- Chiang, C.; Swan, R.Z.; Grachtchouk, M.; Bolinger, M.; Litingtung, Y.; Robertson, E.K.; Cooper, M.K.; Gaffield, W.; Westphal, H.; Beachy, P.A.; et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev. Biol. 1999, 205, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mill, P. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003, 17, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Woo, W.-M.; Zhen, H.H.; Oro, A.E. Shh maintains dermal papilla identity and hair morphogenesis via a Noggin-Shh regulatory loop. Genes Dev. 2012, 26, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Biernaskie, J.; Paris, M.; Morozova, O.; Fagan, B.M.; Marra, M.; Pevny, L.; Miller, F.D. SKPs Derive from Hair Follicle Precursors and Exhibit Properties of Adult Dermal Stem Cells. Cell Stem Cell 2009, 5, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Giangreco, A.; Jensen, K.B.; Mulder, K.W.; Watt, F.M. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development (Camb. Engl.) 2009, 136, 2815–2823. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Mascre, G.; Youseff, K.K.; Harel, I.; Michaux, C.; De Geest, N.; Szpalski, C.; Achouri, Y.; Bloch, W.; Hassan, B.A.; et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J. Cell Biol. 2009, 187, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Williams, J.S.; Brownell, I. Merkel cells and touch domes: More than mechanosensory functions? Exp. Dermatol. 2014, 23, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Thoresen, D.T.; Miao, L.; Williams, J.S.; Wang, C.; Atit, R.P.; Wong, S.Y.; Brownell, I. A Cascade of Wnt, Eda, and Shh Signaling Is Essential for Touch Dome Merkel Cell Development. PLoS Genet. 2016, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Dauber, K.L.; Bar, C.; Tsai, P.C.; Valdes, V.J.; Cohen, I.; Santoriello, F.J.; Zhao, D.; Zheng, D.; Hsu, Y.C.; et al. Polycomb-Mediated Repression and Sonic Hedgehog Signaling Interact to Regulate Merkel Cell Specification during Skin Development. PLoS Genet. 2016, 12, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996, 87, 553–563. [Google Scholar] [CrossRef]
- Adolphe, C.; Hetherington, R.; Ellis, T.; Wainwright, B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006, 66, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, E.; Motoyama, J.; Barnfield, P.C.; Yoshikawa, Y.; Zhang, X.; Mo, R.; Crackower, M.A.; Hui, C.-C. Mice with a Targeted Mutation of Patched2 Are Viable but Develop Alopecia and Epidermal Hyperplasia. Mol. Cell. Biol. 2006, 26, 6609–6622. [Google Scholar] [CrossRef] [PubMed]
- Adolphe, C.; Nieuwenhuis, E.; Villani, R.; Li, Z.J.; Kaur, P.; Hui, C.; Wainwright, B.J. Patched 1 and Patched 2 Redundancy Has a Key Role in Regulating Epidermal Differentiation. J. Investig. Dermatol. 2014, 134, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Mill, P.; Mo, R.; Hu, M.C.; Dagnino, L.; Rosenblum, N.D.; Hui, C.-C. Shh controls epithelial proliferation via independent pathways that converge on N-Myc. Dev. Cell 2005, 9, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Adolphe, C.; Junker, J.P.; Lyubimova, A.; van Oudenaarden, A.; Wainwright, B. Patched Receptors Sense, Interpret, and Establish an Epidermal Hedgehog Signaling Gradient. J. Investig. Dermatol. 2017, 137, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, J.K.; Beckers, S.; Zegers, D.; Van Hul, W. Wnt Signaling and the Control of Human Stem Cell Fate. Stem Cell Rev. Rep. 2014, 10, 207–229. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Merrill, B.J.; Jamora, C.; Dasgupta, R. At the Roots of a Never-Ending Cycle. Dev. Cell 2001, 1, 13–25. [Google Scholar] [CrossRef]
- Widelitz, R.B. Wnt signaling in skin organogenesis. Organogenesis 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Andl, T.; Bagasra, A.; Lu, M.M.; Epstein, D.J.; Morrisey, E.E.; Millar, S.E. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech. Dev. 2001, 107, 69–82. [Google Scholar] [CrossRef]
- Kwack, M.H.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Wnt5a attenuates Wnt/β-catenin signalling in human dermal papilla cells. Exp. Dermatol. 2013, 22, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; DasGupta, R.; Kocieniewski, P.; Fuchs, E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 2003, 422, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Rendl, M.; Polak, L.; Fuchs, E. BMP signaling in dermal papilla cells is required for their hair follicle-inductive properties. Genes Dev. 2008, 22, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.E. Molecular mechanisms regulating hair follicle development. J. Investig. Dermatol. 2002, 118, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, M.L.; Millar, S.E. The mammary bud as a skin appendage: Unique and shared aspects of development. J. Mammary Gland Biol. Neoplasia 2006, 11, 187–203. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Paus, R. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nat. Cell Biol. 1999, 1, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Irrthum, A.; Devriendt, K.; Chitayat, D.; Matthijs, G.; Glade, C.; Steijlen, P.M.; Fryns, J.-P.; Van Steensel, M.A.M.; Vikkula, M. Mutations in the Transcription Factor Gene SOX18 Underlie Recessive and Dominant Forms of Hypotrichosis-Lymphedema-Telangiectasia. Am. J. Hum. Genet. 2003, 72, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, D.; Bowles, J.; Nagy, A.; Muscat, G.; Koopman, P. Mice null for sox18 are viable and display a mild coat defect. Mol. Cell. Biol. 2000, 20, 9331–9336. [Google Scholar] [CrossRef] [PubMed]
- Takakura, N.; Yoshida, H.; Kunisada, T.; Nishikawa, S.; Nishikawa, S.I. Involvement of platelet-derived growth factor receptor-alpha in hair canal formation. J. Investig. Dermatol. 1996, 107, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.; Bondjers, C.; Betsholtz, C. Roles for PDGF-A and sonic hedgehog in development of mesenchymal components of the hair follicle. Development (Camb. Engl.) 1999, 126, 2611–2621. [Google Scholar]
- Gao, J.; DeRouen, M.C.; Chen, C.-H.; Nguyen, M.; Nguyen, N.T.; Ido, H.; Harada, K.; Sekiguchi, K.; Morgan, B.A.; Miner, J.H.; et al. Laminin-511 is an epithelial message promoting dermal papilla development and function during early hair morphogenesis. Genes Dev. 2008, 22, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Tanigaki, K.; Han, H.; Hiai, H.; Honjo, T. Notch/RBP-J signaling regulates epidermis/hair fate determination of hair follicular stem cells. Curr. Biol. 2003, 13, 333–338. [Google Scholar] [CrossRef]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Di Vignano, A.T.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hibbs, M.A.; Gard, A.L.; Shylo, N.A.; Yun, K. Genome-wide analysis of N1ICD/RBPJ targets in vivo reveals direct transcriptional regulation of Wnt, SHH, and Hippo pathway effectors by Notch1. Stem Cells 2012, 30, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Ringuette, R.; Atkins, M.; Lagali, P.S.; Bassett, E.A.; Campbell, C.; Mazerolle, C.; Mears, A.J.; Picketts, D.J.; Wallace, V.A. A Notch-Gli2 axis sustains Hedgehog responsiveness of neural progenitors and Müller glia. Dev. Biol. 2016, 411, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Overbeek, P.A. Involvement of a novel Tnf receptor homologue in hair follicle induction. Nat. Genet. 1999, 22, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Eby, M.T.; Sinha, S.; Jasmin, A.; Chaudhary, P.M. The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J. Biol. Chem. 2001, 276, 2668–2677. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, M.L.; Pispa, J.; Pekkanen, M.; Paulin, L.; Nieminen, P.; Kere, J.; Thesleff, I. Ectodysplasin, a protein required for epithelial morphogenesis, is a novel TNF homologue and promotes cell-matrix adhesion. Mech. Dev. 1999, 88, 133–146. [Google Scholar] [CrossRef]
- Schmidt-Ullrich, R.; Tobin, D.J.; Lenhard, D.; Schneider, P.; Paus, R.; Scheidereit, C. NF-kappaB transmits Eda A1/EdaR signalling to activate Shh and cyclin D1 expression, and controls post-initiation hair placode down growth. Development (Camb. Engl.) 2006, 133, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Segre, J.A. Transcriptional control of epidermal specification and differentiation. Curr. Opin. Genet. Dev. 2004, 14, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Byrne, C. The epidermis: Rising to the surface. Curr. Opin. Genet. Dev. 1994, 4, 725–736. [Google Scholar] [CrossRef]
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Horsley, V.; Fuchs, E. Epithelial Stem Cells: Turning over New Leaves. Cell 2007, 128, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Braun, K.M.; Niemann, C.; Jensen, U.B.; Sundberg, J.P.; Silva-Vargas, V.; Watt, F.M. Manipulation of stem cell proliferation and lineage commitment: Visualisation of label-retaining cells in wholemounts of mouse epidermis. Development (Camb. Engl.) 2003, 130, 5241–5255. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Trempus, C.S.; Morris, R.J.; Bortner, C.D.; Cotsarelis, G.; Faircloth, R.S.; Reece, J.M.; Tennant, R.W. Enrichment for living murine keratinocytes from the hair follicle bulge with the cell surface marker CD34. J. Investig. Dermatol. 2003, 120, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Panteleyev, A.A.; Jahoda, C.A.; Christiano, A.M. Hair follicle predetermination. J. Cell Sci. 2001, 114, 3419–3431. [Google Scholar] [PubMed]
- Barker, N.; van Es, J.H.; Jaks, V.; Kasper, M.; Snippert, H.; Toftgård, R.; Clevers, H. Very long-term self-renewal of small intestine, colon, and hair follicles from cycling Lgr5+ve stem cells. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Liu, Z.Y.; Gambardella, L.; Delacour, A.; Shapiro, R.; Yang, J.; Sizing, I.; Rayhorn, P.; Garber, E.A.; Benjamin, C.D.; et al. Conditional disruption of hedgehog signaling pathway defines its critical role in hair development and regeneration. J. Investig. Dermatol. 2000, 114, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Leopold, P.L.; Crystal, R.G. Induction of the hair growth phase in postnatal mice by localized transient expression of Sonic hedgehog. J. Clin. Investig. 1999, 104, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Brownell, I.; Guevara, E.; Bai, C.B.; Loomis, C.A.; Joyner, A.L. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell 2011, 8, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Pasolli, H.A.; Fuchs, E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 2011, 144, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Mayer, J.A.; de la Cruz, D.; Baker, R.E.; Maini, P.K.; Maxson, R.; Chuong, C.-M. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 2008, 451, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Festa, E.; Fretz, J.; Berry, R.; Schmidt, B.; Rodeheffer, M.; Horowitz, M.; Horsley, V. Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell 2011, 146, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Segal, J.P.; Heller, E.; Lien, W.-H.; Chang, C.-Y.; Guo, X.; Oristian, D.S.; Zheng, D.; Fuchs, E. Nfatc1 orchestrates aging in hair follicle stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E4950–E4959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development (Camb. Engl.) 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorgenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Akladios, B.; Mendoza-Reinoso, V.; Samuel, M.S.; Hardeman, E.C.; Khosrotehrani, K.; Key, B.; Beverdam, A. Epidermal YAP2-5SA-ΔC Drives β-Catenin Activation to Promote Keratinocyte Proliferation in Mouse Skin In Vivo. J. Investig. Dermatol. 2017, 137, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Akladios, B.; Mendoza Reinoso, V.; Cain, J.E.; Wang, T.; Lambie, D.L.; Watkins, D.N.; Beverdam, A. Positive regulatory interactions between YAP and Hedgehog signalling in skin homeostasis and BCC development in mouse skin in vivo. PLoS ONE 2017, 12, e0183178. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Flores, E.R. p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. P63 Is a P53 Homologue Required for Limb and Epidermal Morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Bigelow, R.L.H.; Chari, N.S.; Undén, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgård, R.; McDonnell, T.J. Transcriptional Regulation of bcl-2 Mediated by the Sonic Hedgehog Signaling Pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Regl, G.; Kasper, M.; Schnidar, H.; Eichberger, T.; Neill, G.W.; Philpott, M.P.; Esterbauer, H.; Hauser-Kronberger, C.; Frischauf, A.-M.; Aberger, F. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004, 64, 7724–7731. [Google Scholar] [CrossRef] [PubMed]
- Polakowska, R.R.; Piacentini, M.; Bartlett, R.; Goldsmith, L.A.; Haake, A.R. Apoptosis in human skin development: Morphogenesis, periderm, and stem cells. Dev. Dyn. 1994, 199, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Chari, N.S.; Romano, R.A.; Koster, M.I.; Jaks, V.; Roop, D.; Flores, E.R.; Teglund, S.; Sinha, S.; Gruber, W.; Aberger, F.; et al. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell Death Differ. 2013, 20, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Zhang, X.; Liu, L.; Hui, C.-C.; Mainprize, T.G.; Scherer, S.W.; Wainwright, B.; Hogg, D.; Rutka, J.T. Failure of a medulloblastoma-derived mutant of SUFU to suppress WNT signaling. Oncogene 2004, 23, 4577–4583. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-Cell Carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J. Review of the hair follicle origin hypothesis for basal cell Carcinoma. Dermatol. Surg. 2009, 35, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Khavari, P.A. Modelling cancer in human skin tissue. Nat. Rev. Cancer 2006, 6, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Svärd, J.; Heby-Henricson, K.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, A.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wang, J.; Mancianti, M.L.; Epstein, E.H. Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 2011, 19, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Youssef, K.K.; Lapouge, G.; Bouvrée, K.; Rorive, S.; Brohée, S.; Appelstein, O.; Larsimont, J.-C.; Sukumaran, V.; Van de Sande, B.; Pucci, D.; et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol. 2012, 14, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Li, J.R.; Yao, C.Y.; Urman, N.M.; Chang, A.L.S.; Tang, J.Y.; Oro, A.E. Rolling the Genetic Dice: Neutral and Deleterious Smoothened Mutations in Drug-Resistant Basal Cell Carcinoma. J. Investig. Dermatol. 2015, 135, 2138–2141. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, Y.; Tanaka, N. Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. J. Dev. Biol. 2017, 5, 12. https://doi.org/10.3390/jdb5040012
Abe Y, Tanaka N. Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. Journal of Developmental Biology. 2017; 5(4):12. https://doi.org/10.3390/jdb5040012
Chicago/Turabian StyleAbe, Yoshinori, and Nobuyuki Tanaka. 2017. "Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer" Journal of Developmental Biology 5, no. 4: 12. https://doi.org/10.3390/jdb5040012
APA StyleAbe, Y., & Tanaka, N. (2017). Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. Journal of Developmental Biology, 5(4), 12. https://doi.org/10.3390/jdb5040012