
Control of Hedgehog Signalling by the Cilia-Regulated Proteasome

Abstract
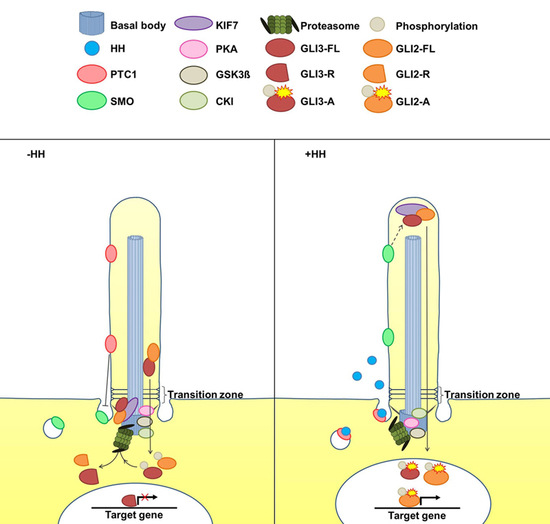
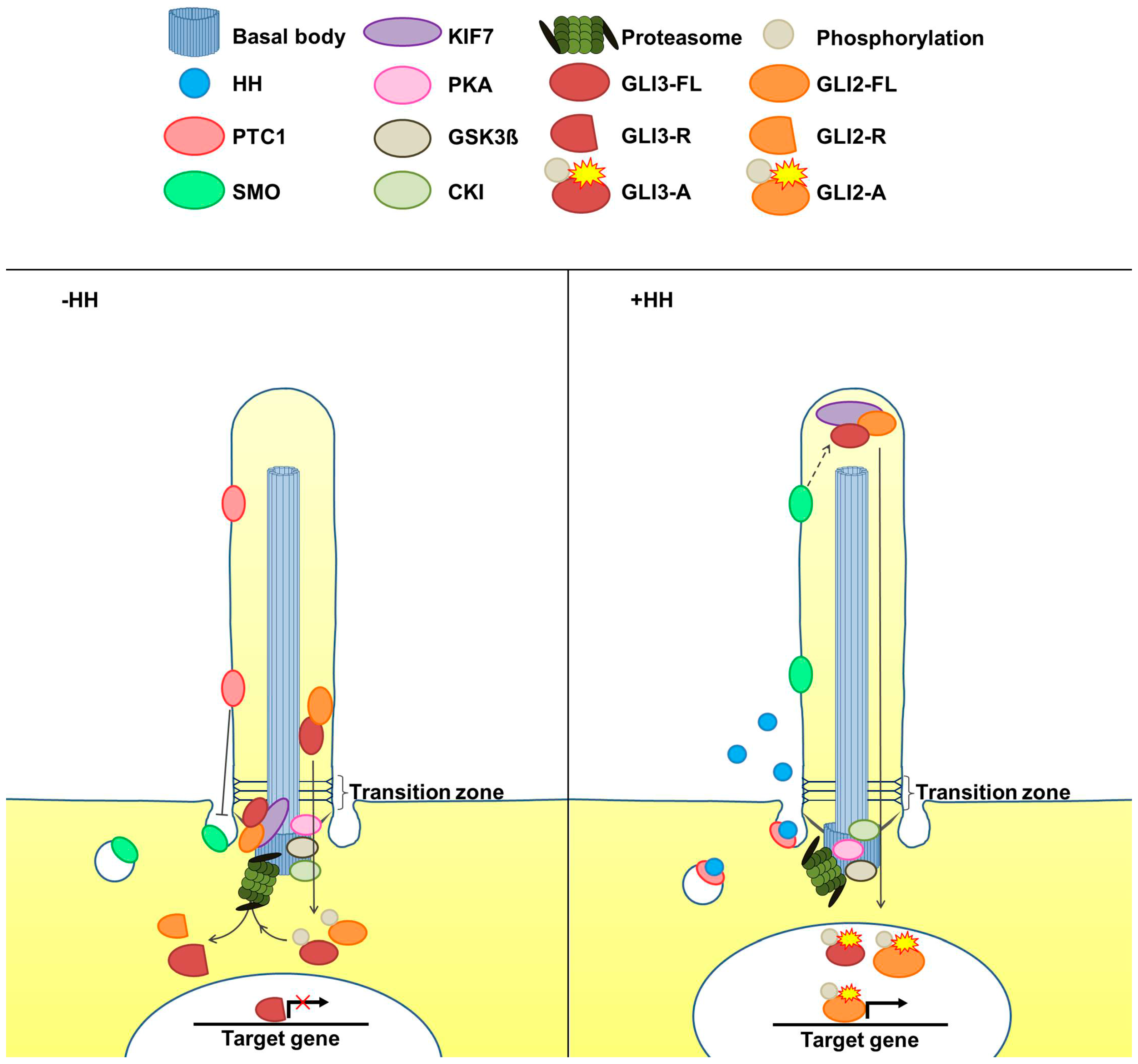
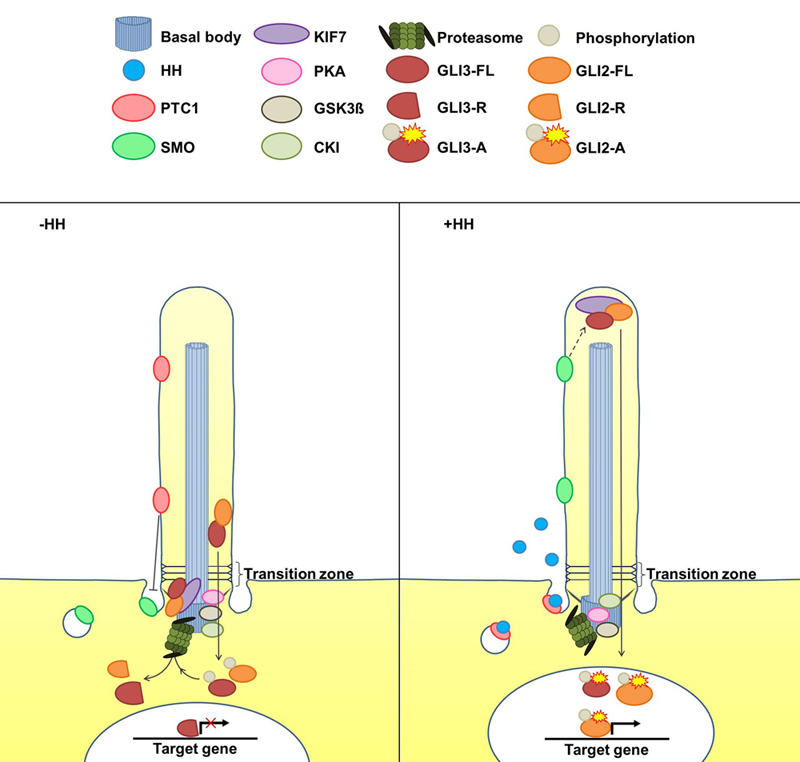{kind=link}
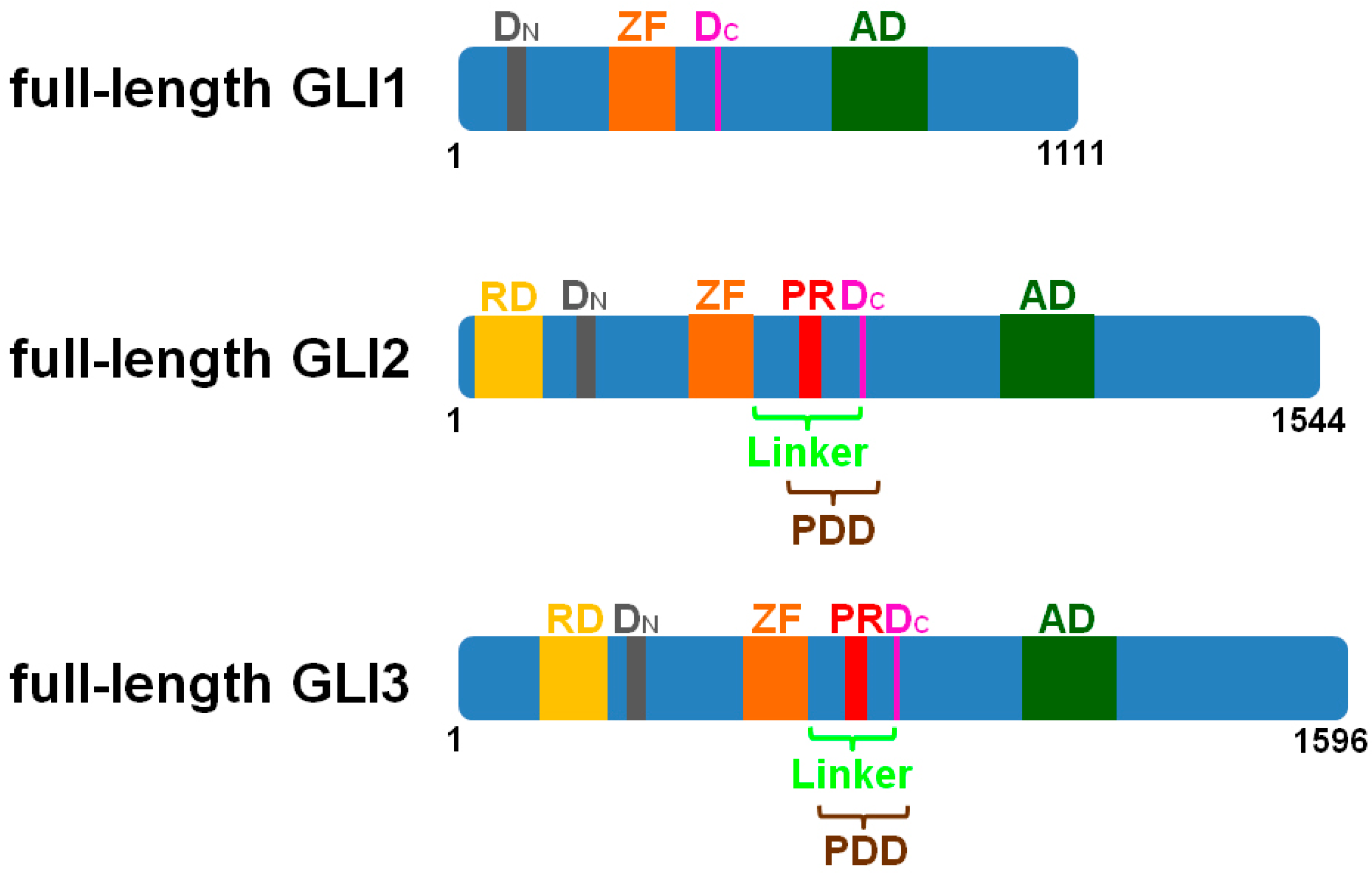{kind=link}
{kind=link}
{kind=link}
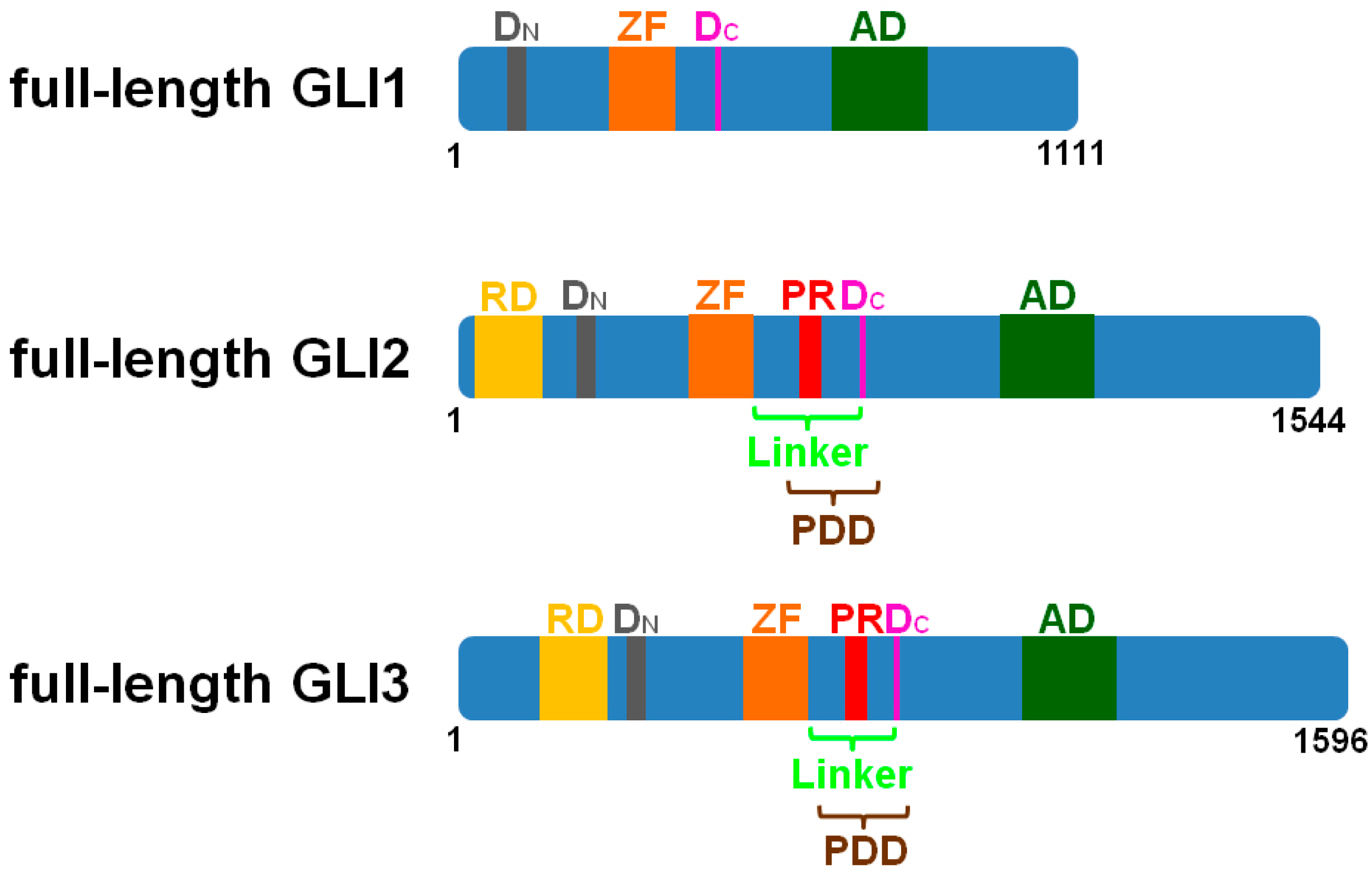
1. Introduction
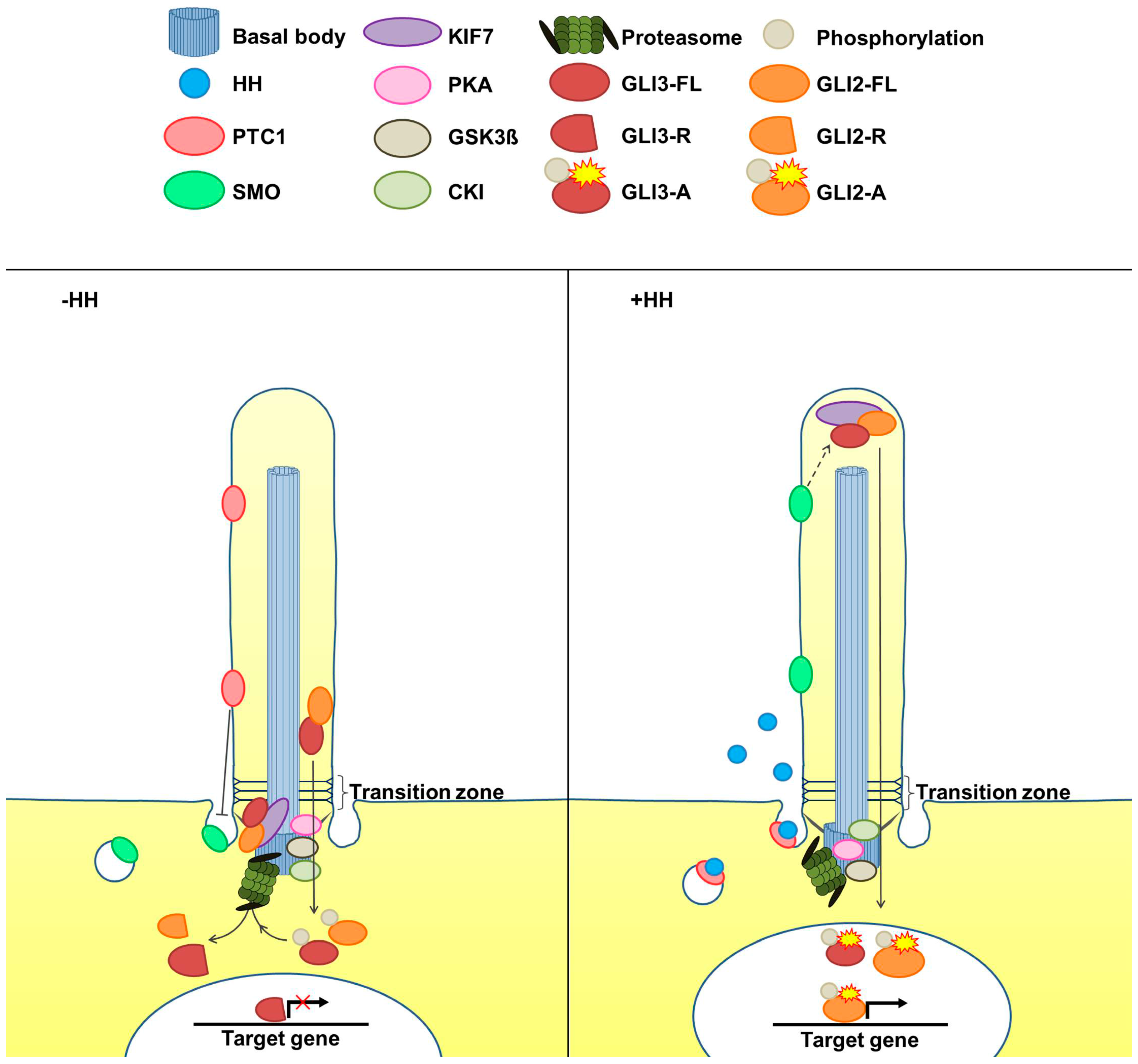
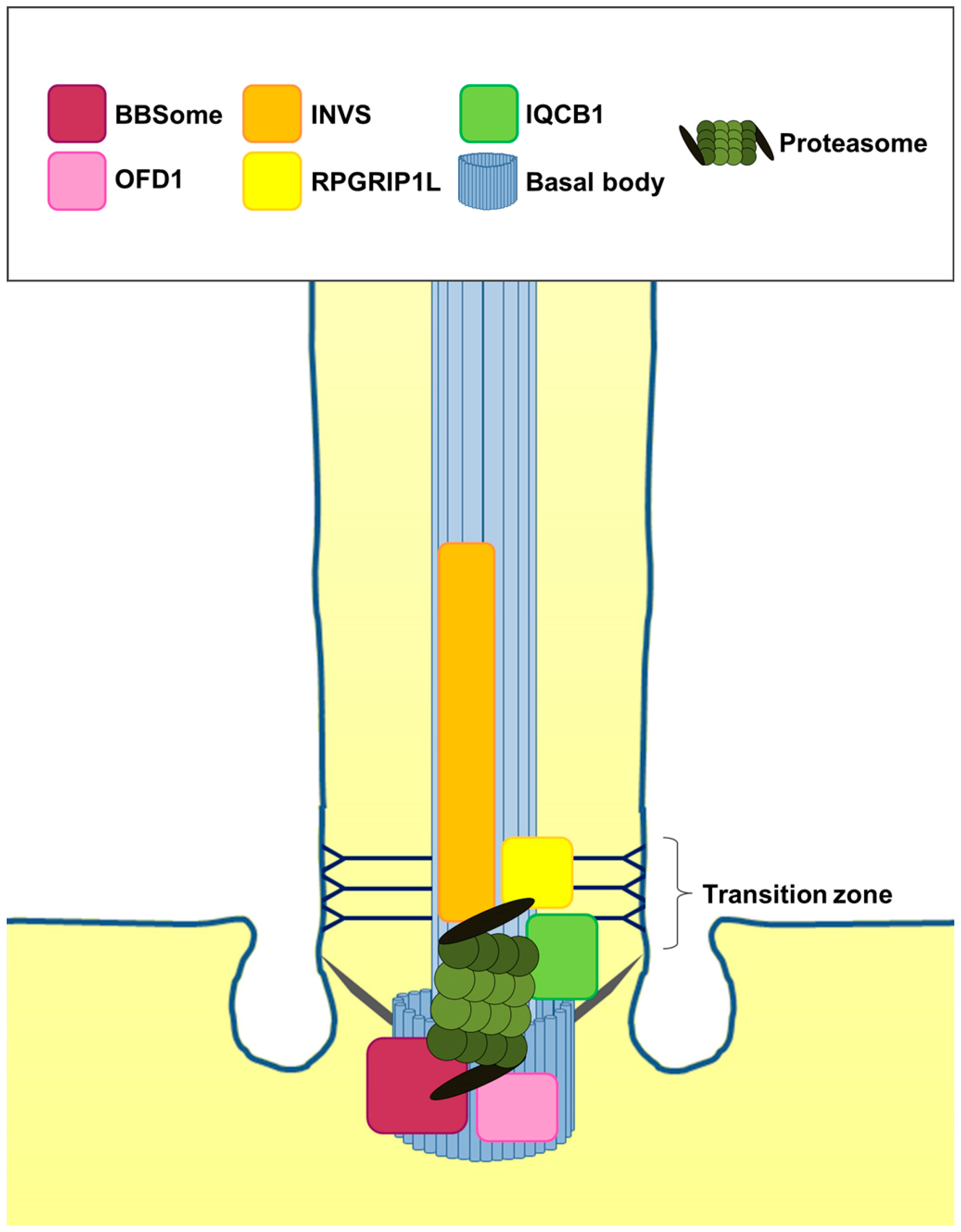
2. The Cilia-Regulated Proteasome Is Essential for GLI Processing
3. GLI2-R and GLI3-R Are Essential for Proper Embryonic Development in Vertebrates
4. Is the Role of the Cilia-Regulated Proteasome Evolutionarily Conserved?
5. Is the Cilia-Regulated Proteasome Involved in the Regulation of PTC and/or SMO Action?
6. Conclusions
- (1)
- Is the proteolytic processing of the GLI2 and GLI3 proteins by the cilia-regulated proteasome evolutionarily conserved?
- (2)
- Does the cilia-regulated proteasome play a role in the control of HH signalling during human embryonic development?
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.; Johnson, R.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.; McMahon, A. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.; Ingham, P. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Ingham, P.; McMahon, A. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Hynes, M.; Stone, D.; Dowd, M.; Pitts-Meek, S.; Goddard, A.; Gurney, A.; Rosenthal, A. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron 1997, 19, 15–26. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A. The works of GLI and the power of hedgehog. Nat. Cell Biol. 1999, 1, E147–E148. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Coux, O.; Tanaka, K.; Goldberg, A. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Fallon, J.; Beachy, P. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, S.; Lüscher, B.; Rüther, U. Transcriptional activity of GLI1 is negatively regulated by protein kinase A. Biol. Chem. 2000, 381, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bai, C.; Platt, K.; Matise, M.; Beeghly, A.; Hui, C.; Nakashima, M.; Joyner, A. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [PubMed]
- Tuson, M.; He, M.; Anderson, K. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Chitalia, V.; Foy, R.; Bachschmid, M.; Zeng, L.; Panchenko, M.; Zhou, M.; Bharti, A.; Seldin, D.; Lecker, S.; Dominguez, I.; et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat. Cell Biol. 2008, 10, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Borgal, L.; Habbig, S.; Hatzold, J.; Liebau, M.; Dafinger, C.; Sacarea, I.; Hammerschmidt, M.; Benzing, T.; Schermer, B. The Ciliary Protein Nephrocystin-4 Translocates the Canonical Wnt-Regulator Jade-1 to the Nucleus to Negatively Regulate Beta-Catenin Signaling. J. Biol. Chem. 2012, 287, 25370–25380. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, T.; Kirschner, M. The master cell cycle regulator APC-Cdc20 regulates ciliary length and disassembly of the primary cilium. eLife 2014, 3, e03083. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.; Joyner, A.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, C.; Pan, Y.; Bai, Z.; Wang, B. Increased proteolytic processing of full-length Gli2 transcription factor reduces the hedgehog pathway activity in vivo. Dev. Dyn. 2011, 240, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Schrader, E.; Harstad, K.; Holmgren, R.; Matouschek, A. A three-part signal governs differential processing of Gli1 and Gli3 proteins by the proteasome. J. Biol. Chem. 2011, 286, 39051–39058. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.; Witman, G. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002, 3, 813–825. [Google Scholar] [CrossRef]
- Goetz, S.; Anderson, K. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.; Anderson, K. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wilson, C.; Li, Y.; Law, K.; Lu, C.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef]
- Humke, E.; Dorn, K.; Milenkovic, L.; Scott, M.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Haycraft, C.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.; Yoder, B. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, T.; Wang, G.; Wang, G.; Chi, W.; Jiang, Q.; Zhang, C. GSK3β-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLoS Biol. 2015, 13, e1002129. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.J.; Kapoor, T.; Anderson, K. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.; Briscoe, J.; Hui, C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Evangelista, M.; Wilson, D.; Wen, X.; Theunissen, J.; Phamluong, K.; Davis, M.; Scales, S.; Solloway, M.; de Sauvage, F.; et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 2009, 19, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Liem, K.J.; He, M.; Ocbina, P.; Anderson, K. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13377–13382. [Google Scholar] [CrossRef] [PubMed]
- Emechebe, U.; Kumar, P.P.; Rozenberg, J.; Moore, B.; Firment, A.; Mirshahi, T.; Moon, A. T-box3 is a ciliary protein and regulates stability of the Gli3 transcription factor to control digit number. eLife 2016, 5, e07897. [Google Scholar] [CrossRef]
- Pedersen, L.; Akhmanova, A. Kif7 keeps cilia tips in shape. Nat. Cell Biol. 2014, 16, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.; Fuertes, G.; Murray, R.; Bose, S.; Knecht, E.; Rechsteiner, M.; Hendil, K.; Tanaka, K.; Dyson, J.; Rivett, J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem. J. 2000, 346, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Lier, J.; Burmühl, S.; Struchtrup, A.; Deutschmann, K.; Vetter, M.; Leu, T.; Reeg, S.; Grune, T.; Rüther, U. The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol. 2015, 210, 115–133. [Google Scholar] [CrossRef]
- Fabunmi, R.; Wigley, W.; Thomas, P.; DeMartino, G. Activity and regulation of the centrosome-associated proteasome. J. Biol. Chem. 2000, 275, 409–413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wigley, W.; Fabunmi, R.; Lee, M.; Marino, C.; Muallem, S.; DeMartino, G.; Thomas, P. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, C.; DeMartino, G. Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 2003, 35, 579–589. [Google Scholar] [CrossRef]
- Amato, R.; Morleo, M.; Giaquinto, L.; di Bernardo, D.; Franco, B. A network-based approach to dissect the cilia/centrosome complex interactome. BMC Genom. 2014, 15, 658. [Google Scholar] [CrossRef]
- Liu, Y.; Tsai, I.; Morleo, M.; Oh, E.; Leitch, C.; Massa, F.; Lee, B.; Parker, D.; Finley, D.; Zaghloul, N.; et al. Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Investig. 2014, 124, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Miller, J.; Corbit, K.; Giles, R.; Brauer, M.; Otto, E.; Baye, L.; Wen, X.; Scales, S.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Leu, T.; Lier, J.; Rüther, U. The cilia-regulated proteasome and its role in the development of ciliopathies and cancer. Cilia 2016, 5, 14. [Google Scholar] [CrossRef]
- Gerdes, J.; Liu, Y.; Zaghloul, N.; Leitch, C.; Lawson, S.; Kato, M.; Beachy, P.; Beales, P.; DeMartino, G.; Fisher, S.; et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007, 39, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Gascue, C.; Tan, P.; Cardenas-Rodriguez, M.; Libisch, G.; Fernandez-Calero, T.; Liu, Y.P.; Astrada, S.; Robello, C.; Naya, H.; Katsanis, N.; et al. Direct role of Bardet-Biedl syndrome proteins in transcriptional regulation. J. Cell Sci. 2012, 125, 362–375. [Google Scholar] [CrossRef]
- Chamling, X.; Seo, S.; Bugge, K.; Searby, C.; Guo, D.; Drack, A.; Rahmouni, K.; Sheffield, V. Ectopic expression of human BBS4 can rescue Bardet-Biedl syndrome phenotypes in Bbs4 null mice. PLoS ONE 2013, 8, e59101. [Google Scholar] [CrossRef] [PubMed]
- Eichers, E.; Abd-El-Barr, M.; Paylor, R.; Lewis, R.; Bi, W.; Lin, X.; Meehan, T.; Stockton, D.; Wu, S.; Lindsay, E.; et al. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum. Genet. 2006, 120, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Mullins, R.; Andrews, M.; Chiang, A.; Swiderski, R.; Yang, B.; Braun, T.; Casavant, T.; Stone, E.; Sheffield, V. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 8664–8669. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; May-Simera, H.; Eichers, E.; Kai, M.; Hill, J.; Jagger, D.; Leitch, C.; Chapple, J.; Munro, P.; Fisher, S.; et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005, 37, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, K.; Fath, M.; Seo, S.; Thedens, D.; Berry, C.; Weiss, R.; Nishimura, D.; Sheffield, V. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J. Clin. Investig. 2008, 118, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Nishimura, D.; Vogel, T.; Shao, J.; Swiderski, R.; Yin, T.; Searby, C.; Carter, C.; Kim, G.; Bugge, K.; et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J. Cell Sci. 2013, 126, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Bimonte, S.; de Angelis, A.; Quagliata, L.; Giusti, F.; Tammaro, R.; Dallai, R.; Ascenzi, M.; Diez-Roux, G.; Franco, B. Ofd1 is required in limb bud patterning and endochondral bone development. Dev. Biol. 2011, 349, 179–191. [Google Scholar] [CrossRef]
- Ferrante, M.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dollé, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Vierkotten, J.; Dildrop, R.; Peters, T.; Wang, B.; Rüther, U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development 2007, 134, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, C.; Lier, J.; Kuschel, S.; Rüther, U. The ciliary protein Ftm is required for ventricular wall and septal development. PLoS ONE 2013, 8, e57545. [Google Scholar] [CrossRef] [PubMed]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Breusing, N.; Arndt, J.; Voss, P.; Bresgen, N.; Wiswedel, I.; Gardemann, A.; Siems, W.; Grune, T. Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mech. Ageing Dev. 2009, 130, 748–753. [Google Scholar] [CrossRef]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M.; et al. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Hu, Z.; Chen, C.; Chen, Y.; Gucek, M.; Li, Z.; Markey, S. Dysbindin-associated proteome in the p2 synaptosome fraction of mouse brain. J. Proteome Res. 2014, 13, 4567–4580. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Hamazaki, J.; Koike, M.; Hirano, Y.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Murata, S. PAC1 gene knockout reveals an essential role of chaperone-mediated 20S proteasome biogenesis and latent 20S proteasomes in cellular homeostasis. Mol. Cell. Biol. 2010, 30, 3864–3874. [Google Scholar] [CrossRef] [PubMed]
- Mtango, N.; Latham, K. Ubiquitin proteasome pathway gene expression varies in rhesus monkey oocytes and embryos of different developmental potential. Physiol. Genom. 2007, 31, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Shimizu, N.; Tokoro, M.; Nishikawa, S.; Hatanaka, Y.; Anzai, M.; Hamazaki, J.; Kishigami, S.; Saeki, K.; Hosoi, Y.; et al. Mouse zygote-specific proteasome assembly chaperone important for maternal-to-zygotic transition. Biol. Open 2013, 2, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Al-Shami, A.; Jhaver, K.; Vogel, P.; Wilkins, C.; Humphries, J.; Davis, J.; Xu, N.; Potter, D.; Gerhardt, B.; Mullinax, R.; et al. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS ONE 2010, 5, e13654. [Google Scholar] [CrossRef]
- Kim, K.; Adelstein, R.; Kawamoto, S. Isoform-specific proteasomal degradation of Rbfox3 during chicken embryonic development. Biochem. Biophys. Res. Commun. 2014, 450, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Marín de Evsikova, C.; Smith, R.; Hicks, W.; Edwards, M.; Longo-Guess, C.; Li, T.; Naggert, J.; Nishina, P. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum. Mol. Genet. 2011, 20, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Louie, C.; Caridi, G.; Lopes, V.; Brancati, F.; Kispert, A.; Lancaster, M.; Schlossman, A.; Otto, E.; Leitges, M.; Gröne, H.; et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 2010, 42, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Atala, A.; Freeman, M.; Mandell, J.; Beier, D. Juvenile cystic kidneys (jck): A new mouse mutation which causes polycystic kidneys. Kidney Int. 1993, 43, 1081–1085. [Google Scholar] [CrossRef] [Green Version]
- Kamat, A.; Karashima, T.; Davis, D.; Lashinger, L.; Bar-Eli, M.; Millikan, R.; Shen, Y.; Dinney, C.; McConkey, D. The proteasome inhibitor bortezomib synergizes with gemcitabine to block the growth of human 253JB-V bladder tumors in vivo. Mol. Cancer Ther. 2004, 3, 279–290. [Google Scholar] [PubMed]
- Papageorgiou, A.; Kamat, A.; Benedict, W.; Dinney, C.; McConkey, D. Combination therapy with IFN-alpha plus bortezomib induces apoptosis and inhibits angiogenesis in human bladder cancer cells. Mol. Cancer Ther. 2006, 5, 3032–3041. [Google Scholar] [CrossRef]
- Konac, E.; Varol, N.; Kiliccioglu, I.; Bilen, C. Synergistic effects of cisplatin and proteasome inhibitor bortezomib on human bladder cancer cells. Oncol. Lett. 2015, 10, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Agyin, J.; Santhamma, B.; Nair, H.; Roy, S.; Tekmal, R. BU-32: A novel proteasome inhibitor for breast cancer. Breast Cancer Res. Treat. 2009, 11, R74. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cuccioloni, M.; Cecarini, V.; Mozzicafreddo, M.; Palermo, F.; Cocci, P.; Angeletti, M.; Eleuteri, A. Ghrelin induces apoptosis in colon adenocarcinoma cells via proteasome inhibition and autophagy induction. Apoptosis 2013, 18, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.; Hedley, D.; Major, P.; Townsley, C.; Mackenzie, M.; Vincent, M.; Degendorfer, P.; Tsao, M.; Nicklee, T.; Birle, D.; et al. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin. Cancer Res. 2005, 11, 5526–5533. [Google Scholar] [CrossRef]
- Ao, L.; Reichel, D.; Hu, D.; Jeong, H.; Kim, K.; Bae, Y.; Lee, W. Polymer Micelle Formulations of Proteasome Inhibitor Carfilzomib for Improved Metabolic Stability and Anti-Cancer Efficacy in Human Multiple Myeloma and Lung Cancer Cell Lines. J. Pharmacol. Exp. Ther. 2015, 355, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, J.; Nie, J.; Sheng, W.; Cao, H.; Shen, W.; Dong, A.; Zhou, J.; Jiao, Y.; Zhang, S.; et al. MG132 enhances the radiosensitivity of lung cancer cells in vitro and in vivo. Oncol. Rep. 2015, 34, 2083–2089. [Google Scholar] [CrossRef] [PubMed]
- Bazzaro, M.; Lee, M.; Zoso, A.; Stirling, W.; Santillan, A.; Shih, I.; Roden, R. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006, 66, 3754–3763. [Google Scholar] [CrossRef]
- Mlynarczuk-Bialy, I.; Doeppner, T.; Golab, J.; Nowis, D.; Wilczynski, G.; Parobczak, K.; Wigand, M.; Hajdamowicz, M.; Biały, L.; Aniolek, O.; et al. Biodistribution and Efficacy Studies of the Proteasome Inhibitor BSc2118 in a Mouse Melanoma Model. Transl. Oncol. 2014, 7, 570–579. [Google Scholar] [CrossRef]
- Bold, R.; Virudachalam, S.; McConkey, D. Chemosensitization of pancreatic cancer by inhibition of the 26S proteasome. J. Surg. Res. 2001, 100, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Befani, C.; Vlachostergios, P.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.; Liakos, P. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. (Berl.) 2012, 90, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.; Taber, D.; Ansari, R.; Ryan, C.; George, C.; Vokes, E.; Vogelzang, N.; Stadler, W. Phase II trial of PS-341 in patients with renal cell cancer: A University of Chicago phase II consortium study. J. Clin. Oncol. 2004, 22, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Kondagunta, G.; Drucker, B.; Schwartz, L.; Bacik, J.; Marion, S.; Russo, P.; Mazumdar, M.; Motzer, R. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J. Clin. Oncol. 2004, 22, 3720–3725. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, S.; Grabowski, D.; Hill, J.; Rybicki, L.; Burk, R.; Bukowski, R.; Ganapathi, M.; Ganapathi, R. Inhibition of proteasome activity by bortezomib in renal cancer cells is p53 dependent and VHL independent. Anticancer Res. 2009, 29, 2961–2969. [Google Scholar] [PubMed]
- Vlashi, E.; Kim, K.; Lagadec, C.; Donna, L.; McDonald, J.; Eghbali, M.; Sayre, J.; Stefani, E.; McBride, W.; Pajonk, F. In vivo imaging, tracking, and targeting of cancer stem cells. J. Natl. Cancer Inst. 2009, 101, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Chan, M.; Frohnen, P.; McDonald, A.; Pajonk, F. Targeted elimination of breast cancer cells with low proteasome activity is sufficient for tumor regression. Breast Cancer Res. Treat. 2013, 141, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Korkaya, H.; Liu, S.; Lee, H.; Newman, B.; Yu, Y.; Clouthier, S.; Schwartz, S.; Wicha, M.; et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin. Cancer Res. 2010, 16, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T. Targeting cancer stem cells with sulforaphane, a dietary component from broccoli and broccoli sprouts. Future Oncol. 2013, 9, 1097–1103. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Bhuta, S.; Lai, C.; Mischel, P.; Werner, M.; Henke, M.; Pajonk, F. Tumor cells with low proteasome subunit expression predict overall survival in head and neck cancer patients. BMC Cancer 2014, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fu, J.; Watkins, D.; Srivastava, R.; Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol. Cell. Biochem. 2013, 373, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Komada, M.; Saitsu, H.; Shiota, K.; Ishibashi, M. Expression of Fgf15 is regulated by both activator and repressor forms of Gli2 in vitro. Biochem. Biophys. Res. Commun. 2008, 369, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Kondrichin, I.; Gong, Z.; Korzh, V. Combined activity of the two Gli2 genes of zebrafish play a major role in Hedgehog signaling during zebrafish neurodevelopment. Mol. Cell. Neurosci. 2008, 37, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Bowers, M.; Eng, L.; Lao, Z.; Turnbull, R.; Bao, X.; Riedel, E.; Mackem, S.; Joyner, A. Limb anterior-posterior polarity integrates activator and repressor functions of GLI2 as well as GLI3. Dev. Biol. 2012, 370, 110–124. [Google Scholar] [CrossRef]
- Wang, C.; Rüther, U.; Wang, B. The Shh-independent activator function of the full-length Gli3 protein and its role in vertebrate limb digit patterning. Dev. Biol. 2007, 305, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Choi, S.; Litingtung, Y.; Chiang, C. Sonic hedgehog signaling regulates Gli3 processing, mesenchymal proliferation, and differentiation during mouse lung organogenesis. Dev. Biol. 2004, 270, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Galtseva, A.; Chen, L.; Mo, R.; Hui, C.; Rosenblum, N. Kif3a controls murine nephron number via GLI3 repressor, cell survival, and gene expression in a lineage-specific manner. PLoS ONE 2013, 8, e65448. [Google Scholar] [CrossRef] [PubMed]
- Cain, J.; Islam, E.; Haxho, F.; Chen, L.; Bridgewater, D.; Nieuwenhuis, E.; Hui, C.; Rosenblum, N. GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE 2009, 4, e7313. [Google Scholar] [CrossRef] [PubMed]
- Cain, J.; Islam, E.; Haxho, F.; Blake, J.; Rosenblum, N. GLI3 repressor controls functional development of the mouse ureter. J. Clin. Investig. 2011, 121, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sun, L.; Veltmaat, J. Hedgehog and Gli signaling in embryonic mammary gland development. J. Mammary Gland Biol. Neoplasia 2013, 18, 133–138. [Google Scholar] [CrossRef]
- Bok, J.; Dolson, D.; Hill, P.; Rüther, U.; Epstein, D.; Wu, D. Opposing gradients of Gli repressor and activators mediate Shh signaling along the dorsoventral axis of the inner ear. Development 2007, 134, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Böse, J.; Grotewold, L.; Rüther, U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum. Mol. Genet. 2002, 11, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Besse, L.; Neti, M.; Anselme, I.; Gerhardt, C.; Rüther, U.; Laclef, C.; Schneider-Maunoury, S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 2011, 138, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Laclef, C.; Anselme, I.; Besse, L.; Catala, M.; Palmyre, A.; Baas, D.; Paschaki, M.; Pedraza, M.; Métin, C.; Durand, B.; et al. The role of primary cilia in corpus callosum formation is mediated by production of the Gli3 repressor. Hum. Mol. Genet. 2015, 24, 4997–5014. [Google Scholar] [CrossRef] [PubMed]
- Basto, R.; Lau, J.; Vinogradova, T.; Gardiol, A.; Woods, C.; Khodjakov, A.; Raff, J. Flies without centrioles. Cell 2006, 125, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Tong, C.; Wang, G.; Wang, B.; Jia, J.; Jiang, J. Hedgehog-regulated Costal2-kinase complexes control phosphorylation and proteolytic processing of Cubitus interruptus. Dev. Cell 2005, 8, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef]
- Chen, Y.; Gallaher, N.; Goodman, R.; Smolik, S. Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc. Natl. Acad. Sci. USA 1998, 95, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Kalderon, D. Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development 1999, 126, 4331–4339. [Google Scholar] [PubMed]
- Wang, G.; Wang, B.; Jiang, J. Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev. 1999, 13, 2828–2837. [Google Scholar] [CrossRef] [PubMed]
- Aza-Blanc, P.; Ramírez-Weber, F.; Laget, M.; Schwartz, C.; Kornberg, T. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 1997, 89, 1043–1053. [Google Scholar] [CrossRef]
- Wang, Q.; Holmgren, R. The subcellular localization and activity of Drosophila cubitus interruptus are regulated at multiple levels. Development 1999, 126, 5097–5106. [Google Scholar] [PubMed]
- Wang, G.; Jiang, J. Multiple Cos2/Ci interactions regulate Ci subcellular localization through microtubule dependent and independent mechanisms. Dev. Biol. 2004, 268, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Keil, T. Functional morphology of insect mechanoreceptors. Microsc. Res. Tech. 1997, 39, 506–531. [Google Scholar] [CrossRef]
- Fuller, M. Spermatogenesis. In Development of Drosophila; Bate, M., Martinez Arias, A., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1993; Volume 2, pp. 71–147. [Google Scholar]
- Kuzhandaivel, A.; Schultz, S.; Alkhori, L.; Alenius, M. Cilia-mediated hedgehog signaling in Drosophila. Cell Rep. 2014, 7, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Méthot, N.; Basler, K. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development 2001, 128, 733–742. [Google Scholar] [PubMed]
- Rink, J.; Gurley, K.; Elliott, S.; Sánchez Alvarado, A. Planarian Hh signaling regulates regeneration polarity and links Hh pathway evolution to cilia. Science 2009, 326, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Diener, D.; Rosenbaum, J. The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J. Cell Biol. 2009, 186, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chinchilla, P.; Fombonne, J.; Ho, L.; Guix, C.; Keen, J.; Mehlen, P.; Riobo, N. Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell. Biol. 2014, 34, 3855–3866. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Y.; Shi, Q.; Yue, T.; Wang, B.; Jiang, J. Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 2012, 10, e1001239. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Jia, H.; Fan, J.; Liu, Y.; Jia, J. USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol. 2012, 10, e1001238. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Jiang, K.; Liu, Y.; Jia, J. Hrs promotes ubiquitination and mediates endosomal trafficking of smoothened in Drosophila hedgehog signaling. PLoS ONE 2013, 8, e79021. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Mao, F.; Lv, X.; Zhang, Z.; Fu, L.; Lu, Y.; Wu, W.; Zhou, Z.; Zhang, L.; Zhao, Y. Drosophila Vps36 regulates Smo trafficking in Hedgehog signaling. J. Cell Sci. 2013, 126, 4230–4238. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Z.; Zhang, C.; Lv, X.; Zheng, X.; Chen, Z.; Sun, L.; Wang, H.; Zhu, Y.; Zhang, J.; et al. Activation of Smurf E3 ligase promoted by smoothened regulates hedgehog signaling through targeting patched turnover. PLoS Biol. 2013, 11, e1001721. [Google Scholar] [CrossRef]
- Lee, R.; Zhao, Z.; Ingham, P. Hedgehog signalling. Development 2016, 143, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.; Beales, P. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio, E.; Walterhouse, D.; Iannaccone, P. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef]
- Wheatley, D. Primary cilia in normal and pathological tissues. Pathobiology 1995, 63, 222–238. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerhardt, C.; Wiegering, A.; Leu, T.; Rüther, U. Control of Hedgehog Signalling by the Cilia-Regulated Proteasome. J. Dev. Biol. 2016, 4, 27. https://doi.org/10.3390/jdb4030027
Gerhardt C, Wiegering A, Leu T, Rüther U. Control of Hedgehog Signalling by the Cilia-Regulated Proteasome. Journal of Developmental Biology. 2016; 4(3):27. https://doi.org/10.3390/jdb4030027
Chicago/Turabian StyleGerhardt, Christoph, Antonia Wiegering, Tristan Leu, and Ulrich Rüther. 2016. "Control of Hedgehog Signalling by the Cilia-Regulated Proteasome" Journal of Developmental Biology 4, no. 3: 27. https://doi.org/10.3390/jdb4030027
APA StyleGerhardt, C., Wiegering, A., Leu, T., & Rüther, U. (2016). Control of Hedgehog Signalling by the Cilia-Regulated Proteasome. Journal of Developmental Biology, 4(3), 27. https://doi.org/10.3390/jdb4030027