
Roles of Antioxidative Enzymes in Wound Healing

Abstract
:1. Introduction
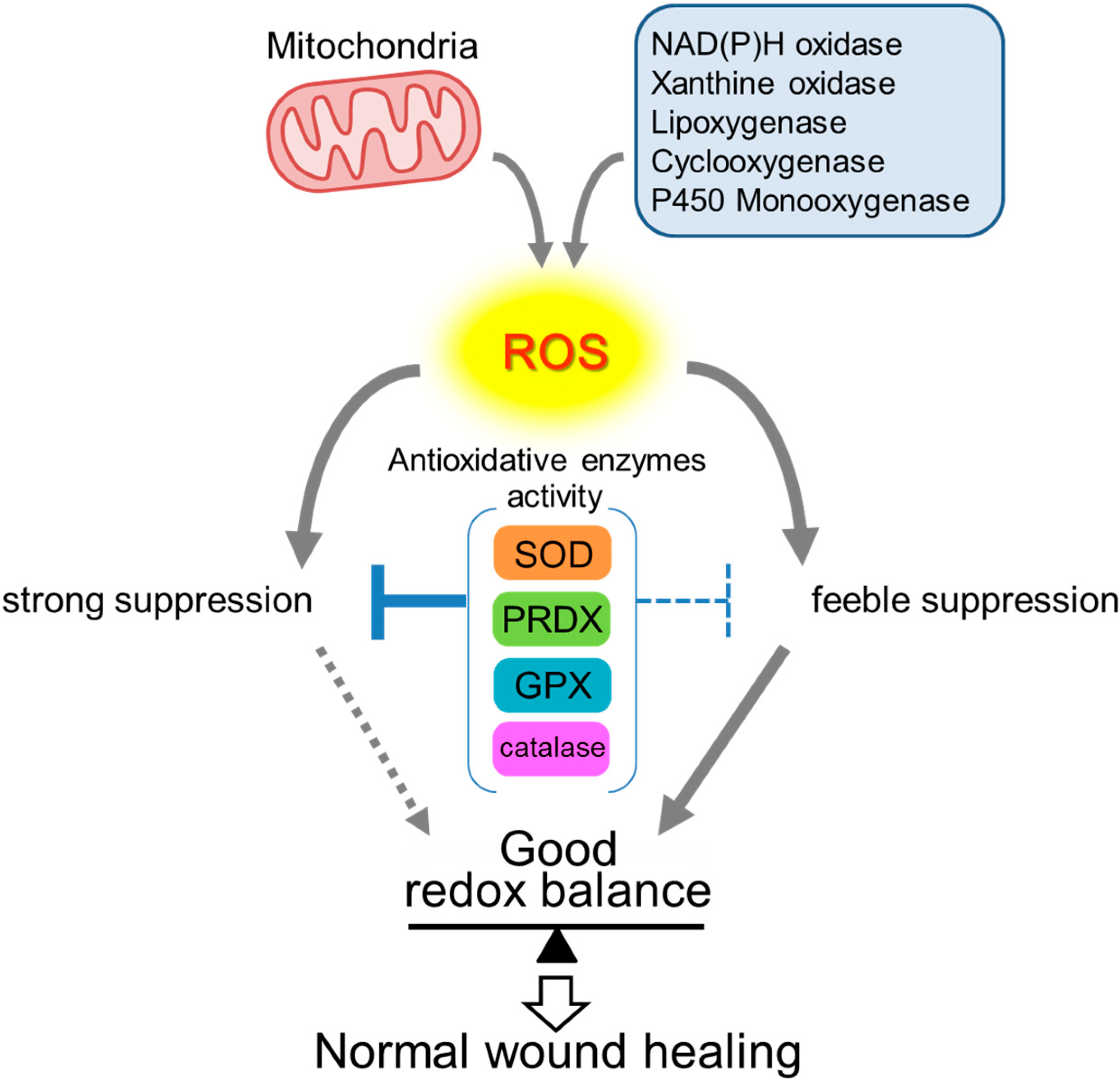
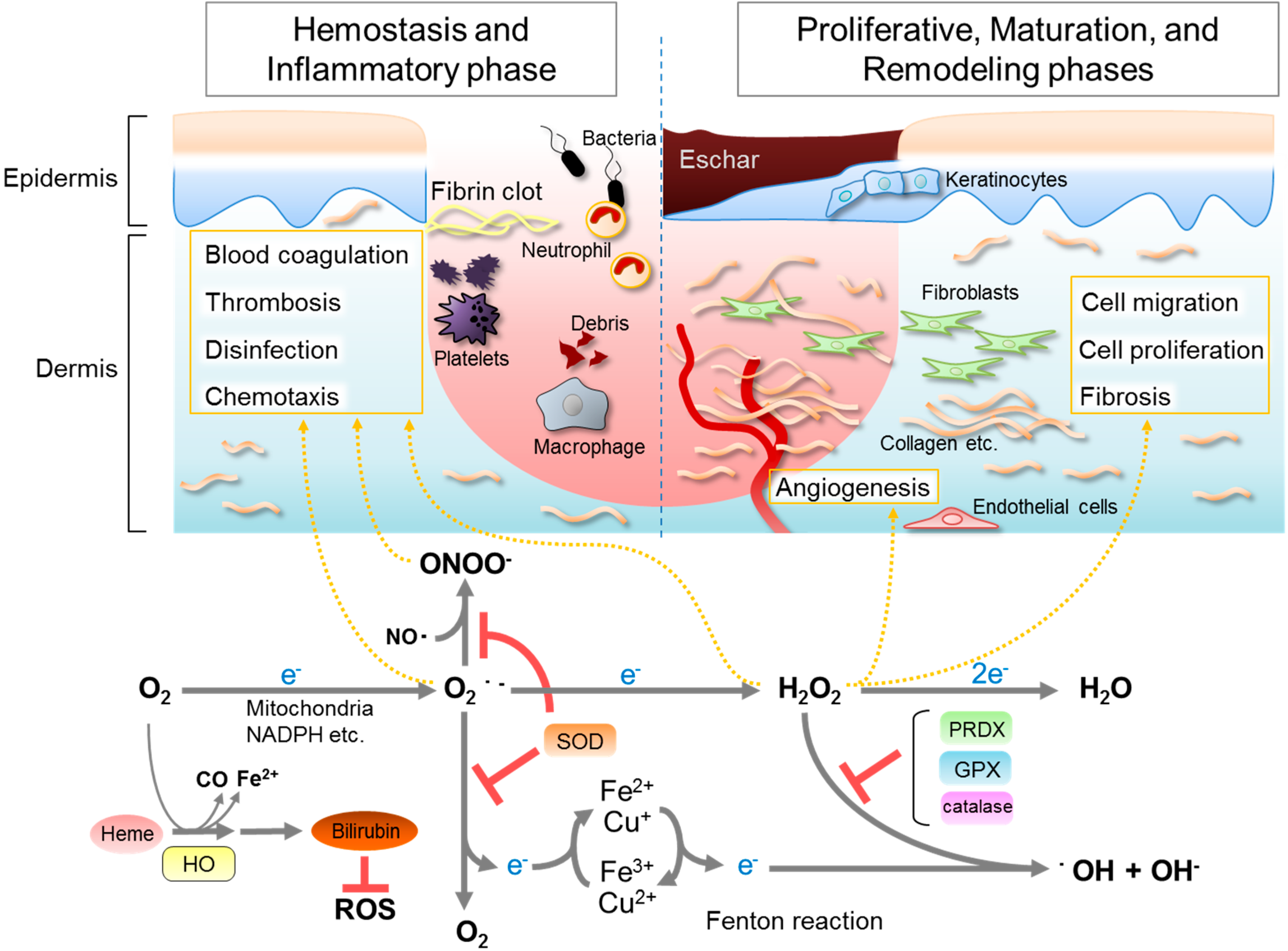
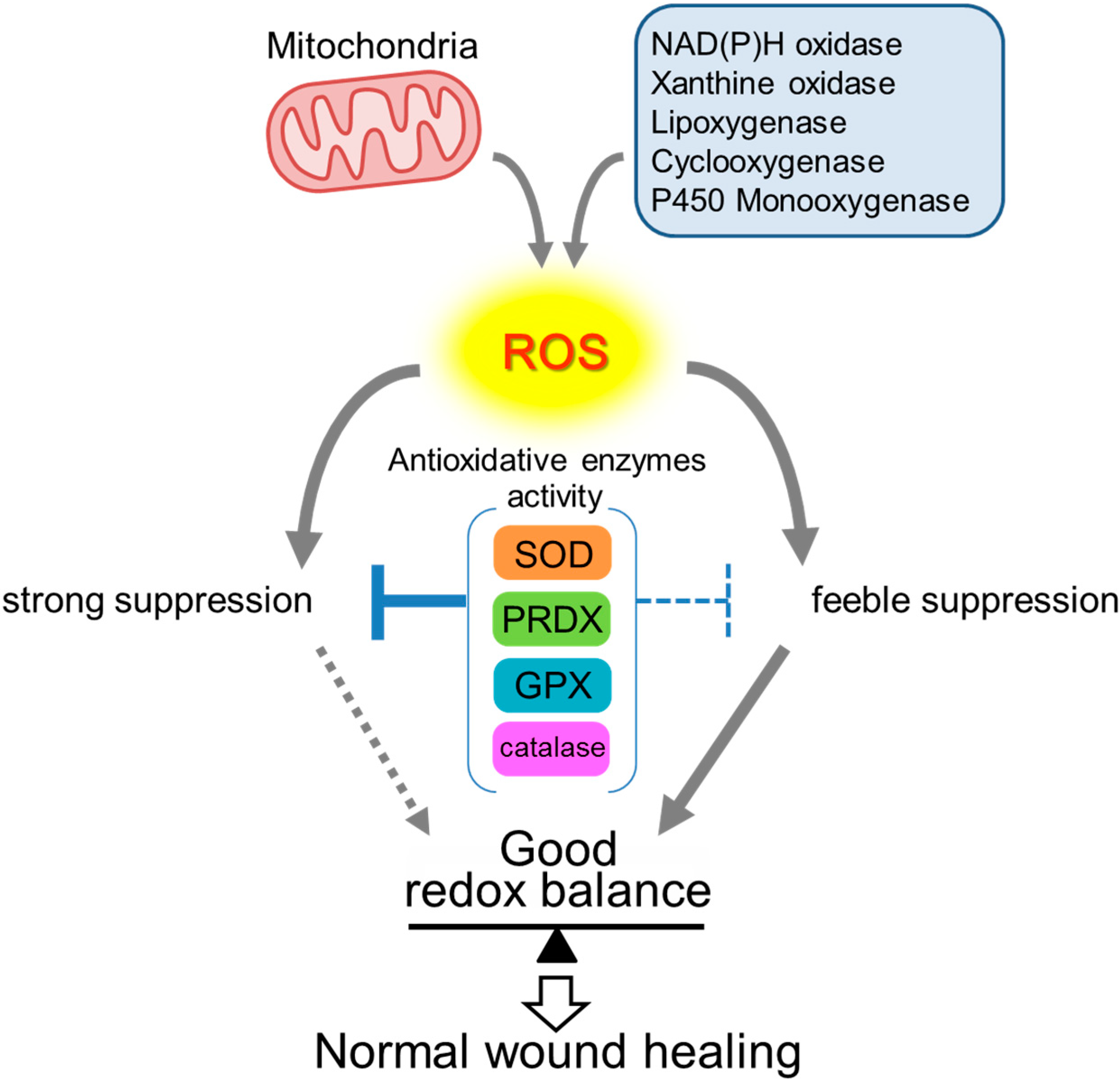
2. Roles of ROS in Wound Healing
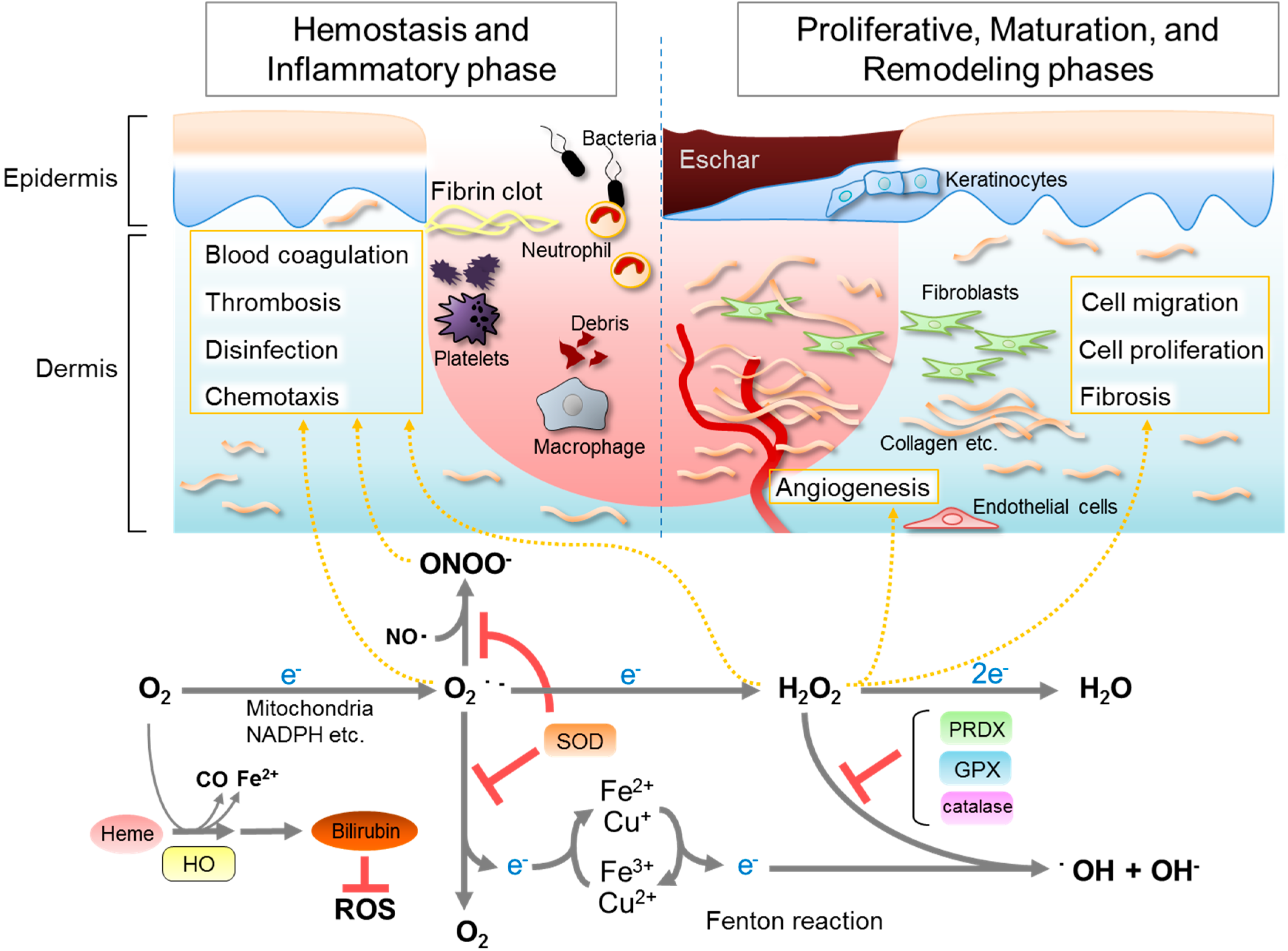
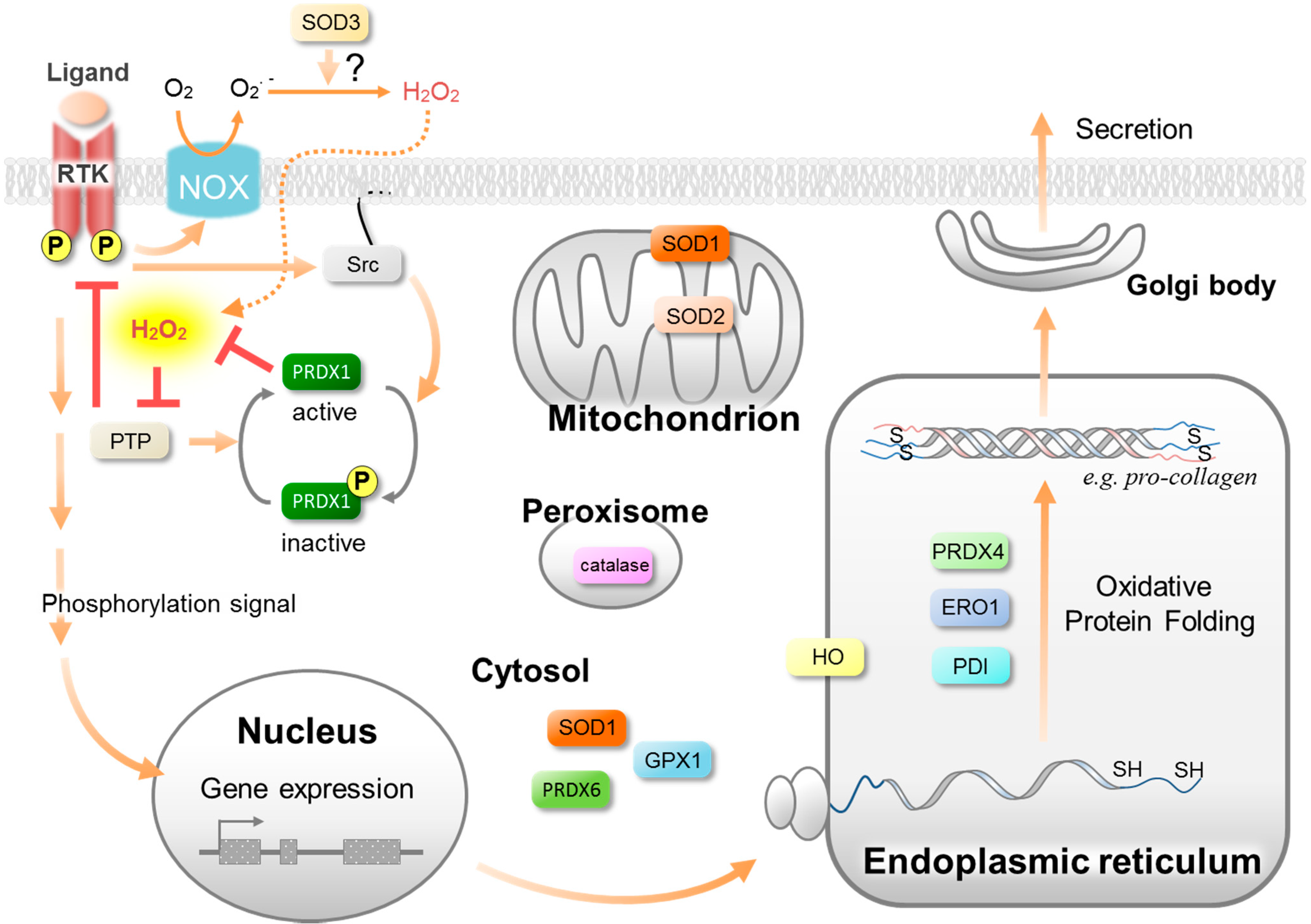
3. Antioxidative Enzymes and Their Roles in Wound Healing
3.1. Superoxide Dismutases (SOD)
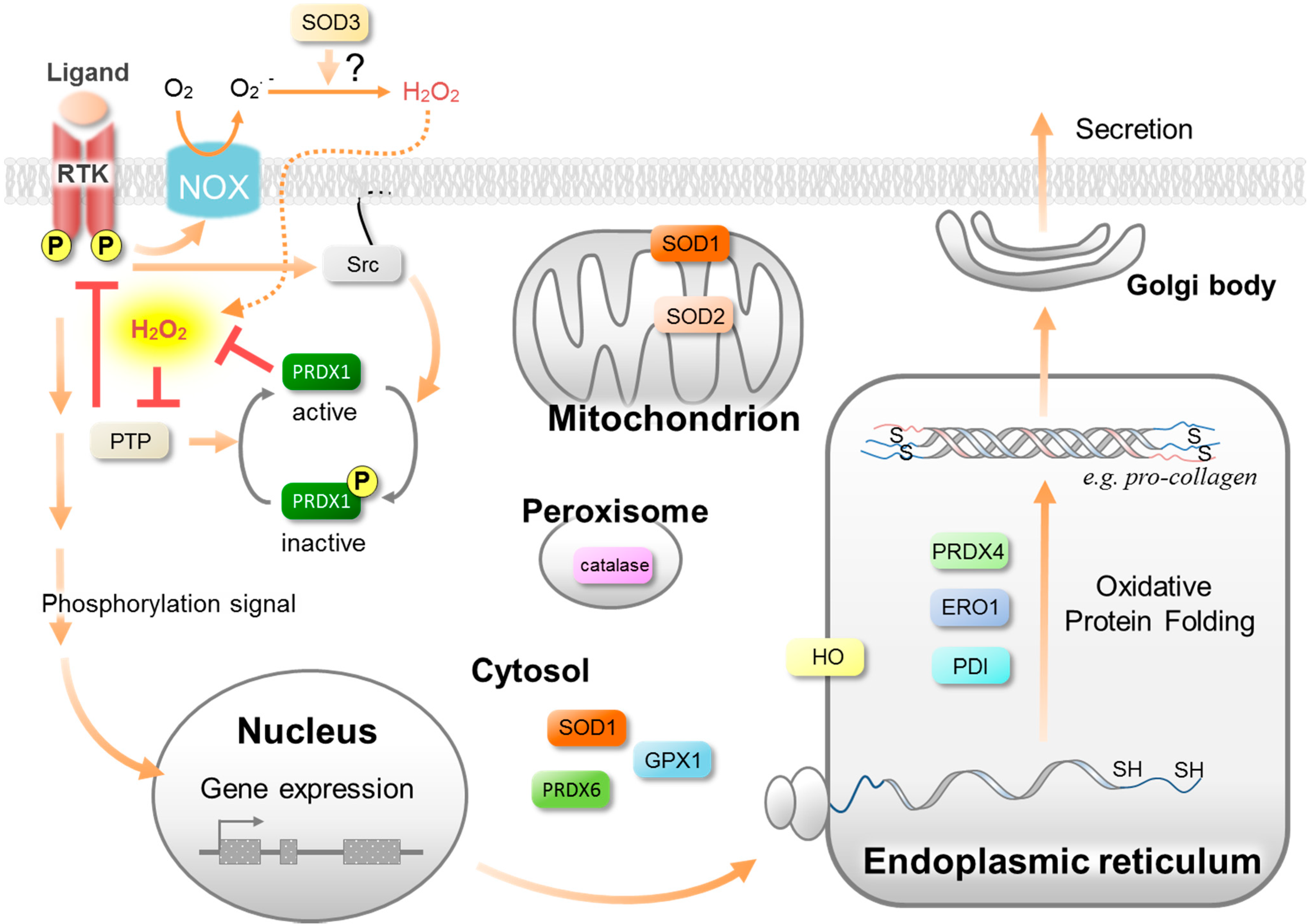
3.2. Peroxiredoxins (PRDX)
3.3. Glutathione Peroxidase (GPX)
3.4. Catalase
3.5. Heme Oxygenases (HO)
4. Conclusions

{kind=link}
{kind=link}
{kind=link}
| Antioxidative Enzyme | Wound Healing Related Phenotype | Reference |
|---|---|---|
| SOD1 | Delayed wound healing at age 20 weeks in KO mice. | [29] |
| SOD2 | Adenovirus-mediated gene therapy of SOD2 rescues the delayed healing in streptozotocin-induced type I diabetes mice. | [38] |
| PRDX6 | Severe hemorrhage in the granulation tissue in KO mice. | [49] |
| Wound closure is enhanced in aged transgenic mice overexpressing PRDX6 in keratinocytes compared with aged wild-type mice. | [50] | |
| Catalase | Impaired wound healing in adenovirus-mediated overexpressing mice. | [16] |
| HO-1 | Impaired re-epithelialization and angiogenesis, resulting in delayed wound healing in KO mice. | [62] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ruth, A.; Bryant, D.P.N. Acute and Chronic Wounds, 4th ed.; Elsevier Inc.: St. Louis, MO, USA, 2010. [Google Scholar]
- Sun, B.K.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen. 2009, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. Redox signals in wound healing. Biochim. Biophys. Acta 2008, 1780, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.D.T.G.; Carels, C.E.; Lundvig, D.M.S. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R. Growth factors and chronic wound healing: Past, present, and future. Adv. Skin Wound Care 2004, 17, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.E.; Parks, W.C. Metalloproteinases and their inhibitors: Regulators of wound healing. Int. J. Biochem. Cell Biol. 2008, 40, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Invest. Dermatol. 1994, 102, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Babior, B.M.; Hunt, T.K.; Ellison, E.C.; Roy, S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem. 2002, 277, 33284–33290. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal wound healing is subject to redox control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Marchese, C.; Maresca, V.; Cardinali, G.; Belleudi, F.; Ceccarelli, S.; Bellocci, M.; Frati, L.; Torrisi, M.R.; Picardo, M. UVB-induced activation and internalization of keratinocyte growth factor receptor. Oncogene 2003, 22, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Goldkorn, T.; Balaban, N.; Matsukuma, K.; Chea, V.; Gould, R.; Last, J.; Chan, C.; Chavez, C. EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am. J. Respir. Cell Mol. Biol. 1998, 19, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Vivekananda, J.; Lin, A.; Coalson, J.J.; King, R.J. Acute inflammatory injury in the lung precipitated by oxidant stress induces fibroblasts to synthesize and release transforming growth factor-alpha. J. Biol. Chem. 1994, 269, 25057–25061. [Google Scholar] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Lee, C.-H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.-Y.; Yang, K.-S.; Lee, S.-R. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Rasik, A.M.; Shukla, A. Antioxidant status in delayed healing type of wounds. Int. J. Exp. Pathol. 2000, 81, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Antioxidants in human health and disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, D.V.; Ankola, D.D.; Bhardwaj, V.; Sahana, D.K.; Kumar, M.N.V.R. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Rasik, A.M.; Patnaik, G.K. Depletion of reduced glutathione, ascorbic acid, vitamin E and antioxidant defence enzymes in a healing cutaneous wound. Free Radic. Res. 1997, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Steiling, H.; Munz, B.; Werner, S.; Brauchle, M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp. Cell Res. 1999, 247, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Roy, D.; Okada, F.; Kibe, N.; Tsunoda, S.; Suzuki, S.; Takahashi, M.; Yokoyama, H.; Yoshitake, J.; Kondo, S.; et al. Spontaneous skin damage and delayed wound healing in SOD1-deficient mice. Mol. Cell. Biochem. 2010, 341, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; de Oliveira, R.M.; Conboy, C.M.; Haigis, M.; Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 2003, 278, 38966–38969. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, E.; Omobono, J.D.; Guo, Z.; Hopkinson, S.; Lazar, A.J.F.; Brenn, T.; Jones, J.C.; Rheinwald, J.G. A keratinocyte hypermotility/growth-arrest response involving laminin 5 and p16INK4A activated in wound healing and senescence. Am. J. Pathol. 2006, 168, 1821–1837. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.; Kibe, N.; Kurahashi, T.; Fujii, J. Differential responses of SOD1-deficient mouse embryonic fibroblasts to oxygen concentrations. Arch. Biochem. Biophys. 2013, 537, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Tamari, Y.; Nawata, H.; Inoue, E.; Yoshimura, A.; Yoshii, H.; Kashino, G.; Seki, M.; Enomoto, T.; Watanabe, M.; Tano, K. Protective roles of ascorbic acid in oxidative stress induced by depletion of superoxide dismutase in vertebrate cells. Free Radic. Res. 2013, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Yao, D.; Grogan, R.H.; Callaghan, M.J.; Edelstein, D.; Brownlee, M.; Gurtner, G.C. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem. 2008, 283, 10930–10938. [Google Scholar] [CrossRef] [PubMed]
- Chiumiento, A.; Lamponi, S.; Barbucci, R.; Domínguez, A.; Pérez, Y.; Villalonga, R. Immobilizing Cu,Zn-superoxide dismutase in hydrogels of carboxymethylcellulose improves its stability and wound healing properties. Biochemistry 2006, 71, 1324–1328. [Google Scholar] [PubMed]
- Luo, J.-D.; Wang, Y.-Y.; Fu, W.-L.; Wu, J.; Chen, A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation 2004, 110, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Ikeda, Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Poole, L.B. Peroxiredoxins as molecular triage agents, sacrificing themselves to enhance cell survival during a peroxide attack. Mol. Cell 2012, 45, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Hansen, H.G.; Yeo, G.S.H.; Fujii, J.; Ron, D. Endoplasmic reticulum thiol oxidase deficiency leads to ascorbic acid depletion and noncanonical scurvy in mice. Mol. Cell 2012, 48, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cells 2010, 40, 787–797. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Kümin, A.; Schäfer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Kümin, A.; Huber, C.; Rülicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006, 169, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, R.L.; Raghubir, R. Antioxidant status during cutaneous wound healing in immunocompromised rats. Mol. Cell. Biochem. 2002, 241, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Fujii, J.; Takao, T.; Kuzuya, T.; Hori, M.; Shimonishi, Y.; Taniguchi, N. The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J. Biol. Chem. 1997, 272, 19152–19157. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Weaver, J.D.; Zhang, S.; Lei, X.G. Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radic. Biol. Med. 2011, 51, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.-J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Blass, S.C.; Goost, H.; Burger, C.; Tolba, R.H.; Stoffel-Wagner, B.; Stehle, P.; Ellinger, S. Extracellular micronutrient levels and pro-/antioxidant status in trauma patients with wound healing disorders: Results of a cross-sectional study. Nutr. J. 2013. [Google Scholar] [CrossRef]
- Nelson, S.M.; Lei, X.; Prabhu, K.S. Selenium levels affect the IL-4-induced expression of alternative activation markers in murine macrophages. J. Nutr. 2011, 141, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Hamilton, H.B.; Neel, J.V.; Kobara, T.Y.; Ogura, Y.; Nishimura, E.T. Hypocatalasemia: A new genetic carrier state. J. Clin. Invest. 1960, 39, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Hanselmann, C.; Mauch, C.; Werner, S. Haem oxygenase-1: A novel player in cutaneous wound repair and psoriasis? Biochem. J. 2001, 353, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Horvitz, H.R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Shyh-Chang, N.; Zhu, H.; de Soysa, T.Y.; Shinoda, G.; Seligson, M.T.; Tsanov, K.M.; Nguyen, L.; Asara, J.M.; Cantley, L.C.; Daley, G.Q. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell 2013, 155, 778–792. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurahashi, T.; Fujii, J. Roles of Antioxidative Enzymes in Wound Healing. J. Dev. Biol. 2015, 3, 57-70. https://doi.org/10.3390/jdb3020057
Kurahashi T, Fujii J. Roles of Antioxidative Enzymes in Wound Healing. Journal of Developmental Biology. 2015; 3(2):57-70. https://doi.org/10.3390/jdb3020057
Chicago/Turabian StyleKurahashi, Toshihiro, and Junichi Fujii. 2015. "Roles of Antioxidative Enzymes in Wound Healing" Journal of Developmental Biology 3, no. 2: 57-70. https://doi.org/10.3390/jdb3020057
APA StyleKurahashi, T., & Fujii, J. (2015). Roles of Antioxidative Enzymes in Wound Healing. Journal of Developmental Biology, 3(2), 57-70. https://doi.org/10.3390/jdb3020057