Epicardium-Derived Heart Repair


Abstract
:1. Cardiovascular Disease
2. Cardiac Regeneration
3. The Epicardium as a Source of Endogenous Progenitor Cells
4. Mouse Models to Identify Epicardial Cells in Vivo
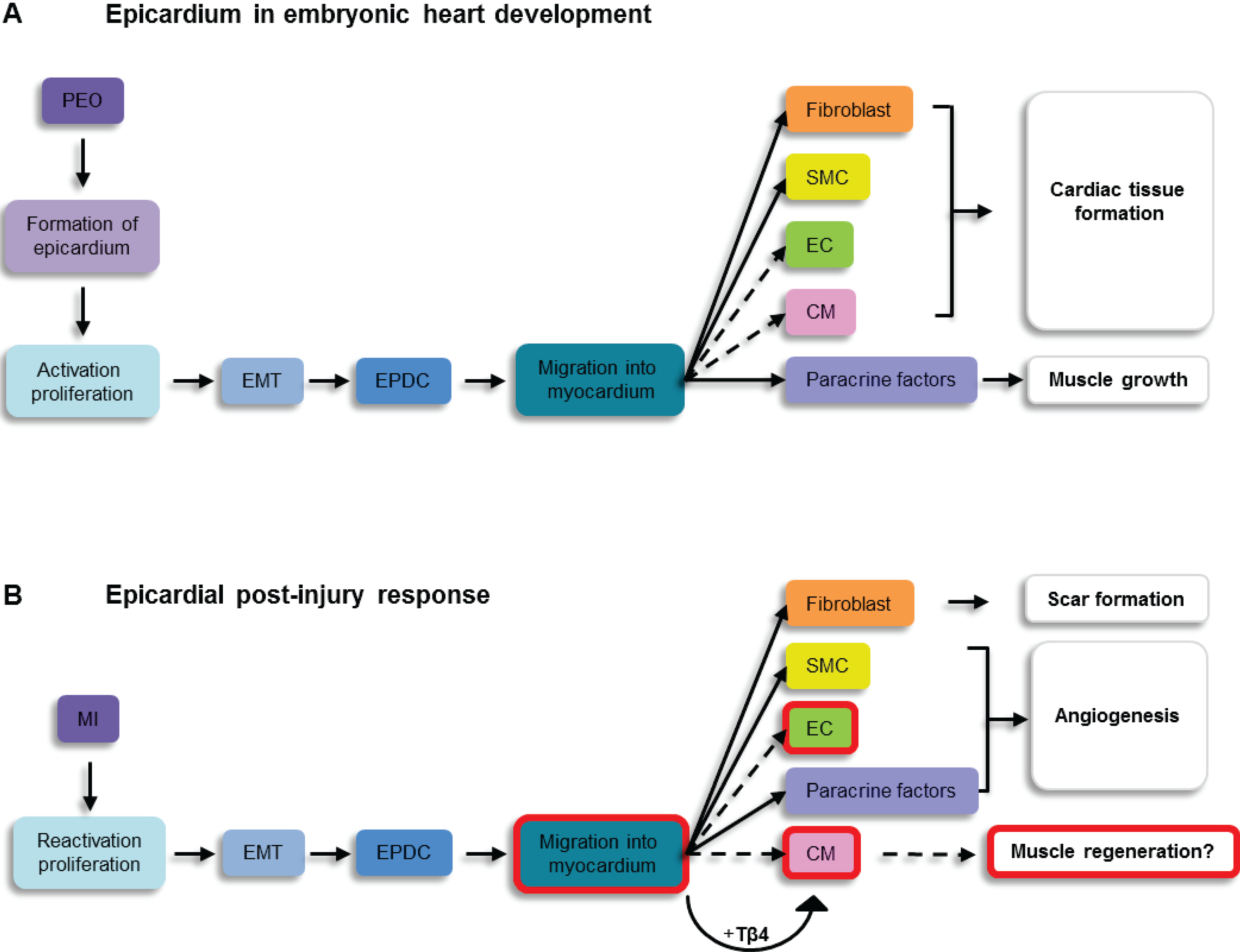
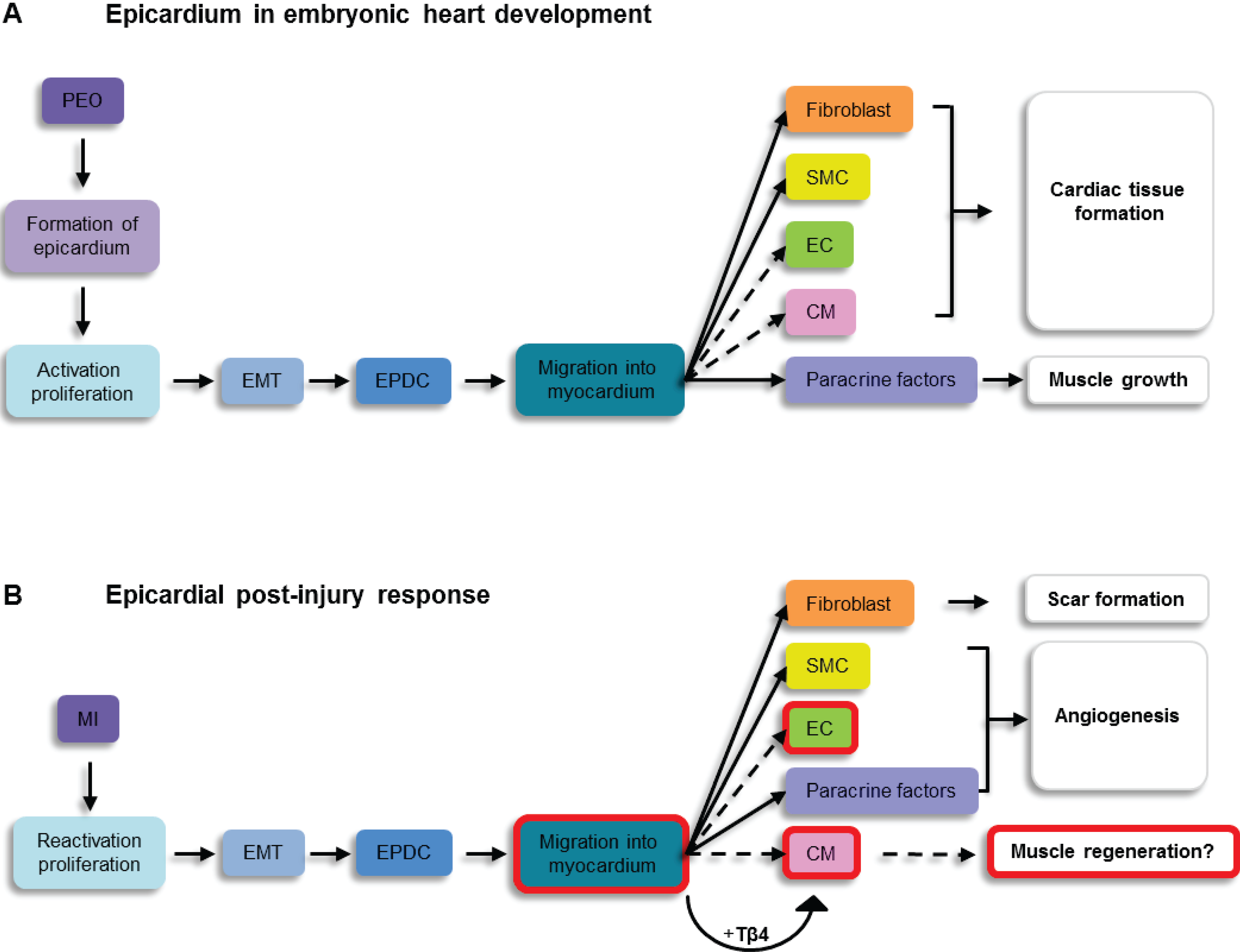
5. The Post-Natal Epicardium
5.1. The Intact Heart
5.2. The Injured Adult Heart
6. Epicardium Derived Cell Differentiation into Cardiovascular Cell Types
6.1. Cardiomyocyte Formation
6.2. Neovascularisation
6.3. Fibrotic Response
7. Discussion

{kind=link}
| Model | Activation | EPDC Differentiation | Markers Used for Identification | Time-points | Details | Reference |
|---|---|---|---|---|---|---|
| WT1CreERT2/+; R26RmTmG | Expansion | SMC Fibro Sporadic EC | SM-MHC, αSMA, SM22α FN1, ColIII, FSP1, ProCol1 PECAM | 14d # | Tamoxifen: twice weekly for 2-3 weeks, MI one week after final injection | 33 |
| Ad:Msln-Cre; R26RmTmG | n.a. | Fibro | FSP1 | 3d-4wks | Ultrasound-guided virus delivery | 33 |
| WT1CreERT2/+; R26REYFP | Expansion Migration | CM* | cTnT, sαActin, Cx43, N-Cad, Ca2+ transients Functional coupling | 14d | Tβ4: Pre- and post-MI Tamoxifen: 5 and 3 days pre- MI | 34 |
| Gata5-Cre; R26REYFP | Expansion Migration | SMC$ EC$ Fibro | αSMA PECAM ProColI | 7d | Tβ4: Pre- and post-MI | 36 |
| WT1CreERT2/+; R26RmTmG | Expansion | SMC Fibro | αSMA DDR2, ProCol1, desmin, FSP1, ColIII | 14d | Tamoxifen: twice weekly for 2-3 weeks, MI one week after final injection Tβ4: Post-MI | 38 |
| (BAC)WT1EGFPCre; R26R | Expansion Migration | SMC EC CM^ Fibro | αSMA PECAM + location in vessel wall cTnI, SERCA2 via exclusion of other markers | 1mo, 3mo (CM) | 43 | |
| LV-CMVGFP | Migration | CM* | sαActin, morphology | 7d, 21d | Sub-pericardial virus injection | 44 |
8. Conclusion
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS. Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Cleutjens, J.P.; Blankesteijn, W.M.; Daemen, M.J.; Smits, J.F. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc. Res. 1999, 44, 232–241. [Google Scholar] [CrossRef]
- Bernstein, H.S.; Srivastava, D. Stem cell therapy for cardiac disease. Pediatr. Res. 2012, 71, 491–499. [Google Scholar] [CrossRef]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; Leri, A.; Anversa, P. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef]
- Balsam, L.B.; Wagers, A.J.; Christensen, J.L.; Kofidis, T.; Weissman, I.L.; Robbins, R.C. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 2004, 428, 668–673. [Google Scholar] [CrossRef]
- Murry, C.E.; Soonpaa, M.H.; Reinecke, H.; Nakajima, H.; Nakajima, H.O.; Rubart, M.; Pasumarthi, K.B.; Virag, J.I.; Bartelmez, S.H.; Poppa, V.; Bradford, G.; Dowell, J.D.; Williams, D.A.; Field, L.J. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004, 428, 664–668. [Google Scholar] [CrossRef]
- Boyle, A.J.; McNiece, I.K.; Hare, J.M. Mesenchymal stem cell therapy for cardiac repair. Methods Mol. Biol. 2010, 660, 65–84. [Google Scholar]
- Assmus, B.; Schachinger, V.; Teupe, C.; Britten, M.; Lehmann, R.; Dobert, N.; Grunwald, F.; Aicher, A.; Urbich, C.; Martin, H.; Hoelzer, D.; Dimmeler, S.; Zeiher, A.M. Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE-AMI). Circulation 2002, 106, 3009–3017. [Google Scholar] [CrossRef]
- Hagege, A.A.; Carrion, C.; Menasche, P.; Vilquin, J.T.; Duboc, D.; Marolleau, J.P.; Desnos, M.; Bruneval, P. Viability and differentiation of autologous skeletal myoblast grafts in ischaemic cardiomyopathy. Lancet 2003, 361, 491–492. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Mummery, C.; Ward-van Oostwaard, D.; Doevendans, P.; Spijker, R.; van den Brink, S.; Hassink, R.; van der Heyden, M.; Opthof, T.; Pera, M.; de la Riviere, A.B.; Passier, R.; Tertoolen, L. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation 2003, 107, 2733–2740. [Google Scholar] [CrossRef]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef]
- Ieda, M.; Fukuda, K. Cardiomyocyte generation using stem cells and directly reprogrammed cells. Front Biosci. (Schol. Ed.) 2012, 4, 1413–1423. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; Jovinge, S.; Frisen, J. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; Leri, A.; Kajstura, J.; Nadal-Ginard, B.; Anversa, P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.; Coletta, M.; Vivarelli, E.; Frati, L.; Cossu, G.; Giacomello, A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfo, M.; Agosti, V.; Viglietto, G.; Condorelli, G.; Indolfi, C.; Ottolenghi, S.; Torella, D.; Nadal-Ginard, B. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef]
- Smith, R.R.; Barile, L.; Cho, H.C.; Leppo, M.K.; Hare, J.M.; Messina, E.; Giacomello, A.; Abraham, M.R.; Marban, E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007, 115, 896–908. [Google Scholar] [CrossRef]
- Smits, A.M.; van Vliet, P.; Metz, C.H.; Korfage, T.; Sluijter, J.P.; Doevendans, P.A.; Goumans, M.J. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomycoytes: An in vitro model for studying human cardiac physiology and pathophysiology. Nat. Protoc. 2009, 4, 232–243. [Google Scholar] [CrossRef]
- Lie-Venema, H.; Van Den Akker, N.M.; Bax, N.A.; Winter, E.M.; Maas, S.; Kekarainen, T.; Hoeben, R.C.; Deruiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Sci. World J. 2007, 7, 1777–1798. [Google Scholar] [CrossRef]
- Reese, D.E.; Mikawa, T.; Bader, D.M. Development of the coronary vessel system. Circ. Res. 2002, 91, 761–768. [Google Scholar] [CrossRef]
- Carmona, R.; Gonzalez-Iriarte, M.; Perez-Pomares, J.M.; Munoz-Chapuli, R. Localization of the Wilm's tumour protein WT1 in avian embryos. Cell Tissue Res. 2001, 303, 173–186. [Google Scholar] [CrossRef]
- Kraus, F.; Haenig, B.; Kispert, A. Cloning and expression analysis of the mouse T-box gene Tbx18. Mech. Dev. 2001, 100, 83–86. [Google Scholar] [CrossRef]
- Moss, J.B.; Xavier-Neto, J.; Shapiro, M.D.; Nayeem, S.M.; McCaffery, P.; Drager, U.C.; Rosenthal, N. Dynamic patterns of retinoic acid synthesis and response in the developing mammalian heart. Dev. Biol. 1998, 199, 55–71. [Google Scholar] [CrossRef]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; Pu, W.T. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; Stallcup, W.B.; Denton, C.P.; McCulloch, A.; Chen, J.; Evans, S.M. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9. [Google Scholar] [CrossRef]
- Rudat, C.; Kispert, A. Wt1 and epicardial fate mapping. Circ. Res. 2012, 111, 165–169. [Google Scholar] [CrossRef]
- Zhou, B.; Pu, W.T. Genetic Cre-loxP Assessment of Epicardial Cell Fate Using Wt1-Driven Cre Alleles. Circ. Res. 2012, 111, e276–e280. [Google Scholar] [CrossRef]
- Wessels, A.; Perez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004, 276, 43–57. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; Gise, A.; Zhou, P.; Hu, Y.W.; Wang, G.; Zhang, B.; Wang, L.; Hall, J.L.; Moses, M.A.; McGowan, F.X.; Pu, W.T. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest 2011, 121, 1894–1904. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; Pu, W.T.; Riley, P.R. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef]
- Nagy, A. Cre recombinase: the universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Clark, J.E.; Ehler, E.; Miquerol, L.; Rossdeutsch, A.; Marber, M.S.; Riley, P.R. Thymosin beta4 facilitates epicardial neovascularization of the injured adult heart. Ann. N. Y. Acad. Sci. 2010, 1194, 97–104. [Google Scholar] [CrossRef]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Nat. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; Ma, Q.; Oh, J.H.; Lin, R.Z.; Melero-Martin, J.M.; von Gise, A.; Zhou, P.; Hu, T.; He, L.; Wu, K.H.; Zhang, H.; Zhang, Y.; Pu, W.T. Thymosin beta 4 treatment after myocardial infarction does not reprogram epicardial cells into cardiomyocytes. J. Mol. Cell Cardiol. 2012, 52, 43–47. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De Mori, R.; Marchetti, C.; Pozzoli, O.; Gentili, C.; Zacheo, A.; Germani, A.; Capogrossi, M.C. Mocardial infarction induces embryonic reprogramming of epicardial c-kit(+) cells: Role of the pericardial fluid. J. Mol. Cell Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef]
- Huang, G.N.; Thatcher, J.E.; McAnally, J.; Kong, Y.; Qi, X.; Tan, W.; DiMaio, J.M.; Amatruda, J.F.; Gerard, R.D.; Hill, J.A.; Bassel-Duby, R.; Olson, E.N. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science 2012, 338, 1599–1603. [Google Scholar] [CrossRef]
- van Wijk, B.; Gunst, Q.D.; Moorman, A.F.; van den Hoff, M.J. Cardiac regeneration from activated epicardium. PLoS ONE 2012, 7, e44692. [Google Scholar]
- Limana, F.; Zacheo, A.; Mocini, D.; Mangoni, A.; Borsellino, G.; Diamantini, A.; De Mori, R.; Battistini, L.; Vigna, E.; Santini, M.; Loiaconi, V.; Pompilio, G.; Germani, A.; Capogrossi, M.C. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ. Res. 2007, 101, 1255–1265. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Poelmann, R.E. Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell Mol. Med. 2010, 14, 1056–1060. [Google Scholar]
- Riley, P.R.; Smart, N. Vascularizing the heart. Cardiovasc. Res. 2011, 91, 260–268. [Google Scholar] [CrossRef]
- Red-Horse, K.; Ueno, H.; Weissman, I.L.; Krasnow, M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [Google Scholar] [CrossRef]
- Tian, X.; Hu, T.; Zhang, H.; He, L.; Huang, X.; Liu, Q.; Yu, W.; He, L.; Yang, Z.; Zhang, Z.; Zhong, T.P.; Yang, X.; Yang, Z.; Yan, Y.; Baldini, A.; Sun, Y.; Lu, J.; Schwartz, R.J.; Evans, S.M.; Gittenberger-de Groot, A.C.; Red-Horse, K.; Zhou, B. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013, 23, 1075–1090. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Bartelings, M.M.; Goumans, M.J.; Deruiter, M.C.; Poelmann, R.E. The arterial and cardiac epicardium in development, disease and repair. Differentiation 2012, 84, 41–53. [Google Scholar] [CrossRef]
- Winter, E.M.; Grauss, R.W.; Hogers, B.; van Tuyn, J.; van der Geest, R.; Lie-Venema, H.; Steijn, R.V.; Maas, S.; Deruiter, M.C.; deVries, A.A.; Steendijk, P.; Doevendans, P.A.; van der Laarse, A.; Poelmann, R.E.; Schalij, M.J.; Atsma, D.E.; Gittenberger-de Groot, A.C. Preservation of left ventricular function and attenuation of remodeling after transplantation of human epicardium-derived cells into the infarcted mouse heart. Circulation 2007, 116, 917–927. [Google Scholar] [CrossRef]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell Cardiol. 2013. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Janardhanan, R.; Salerno, M.; Hunter, J.R.; Burdick, M.M.; Field, J.J.; Strieter, R.M.; Kramer, C.M. Elevated circulating fibrocyte levels in patients with hypertensive heart disease. J. Hypertens. 2012, 30, 1856–1861. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; Neilson, E.G.; Sayegh, M.H.; Izumo, S.; Kalluri, R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; Majesky, M.; Deb, A. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef]
- von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- Reinert, R.B.; Kantz, J.; Misfeldt, A.A.; Poffenberger, G.; Gannon, M.; Brissova, M.; Powers, A.C. Tamoxifen-Induced Cre-loxP Recombination Is Prolonged in Pancreatic Islets of Adult Mice. PLoS ONE 2012, 7, e33529. [Google Scholar]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; Dettman, R.W.; Burch, J.B. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Bondke, A.; Nafz, B.; Flemming, B.; Theres, H.; Scholz, H. The Wilms’ tumor suppressor Wt1 is expressed in the coronary vasculature after myocardial infarction. FASEB J. 2002, 16, 1117–1119. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smits, A.M.; Riley, P.R. Epicardium-Derived Heart Repair. J. Dev. Biol. 2014, 2, 84-100. https://doi.org/10.3390/jdb2020084
Smits AM, Riley PR. Epicardium-Derived Heart Repair. Journal of Developmental Biology. 2014; 2(2):84-100. https://doi.org/10.3390/jdb2020084
Chicago/Turabian StyleSmits, Anke M., and Paul R. Riley. 2014. "Epicardium-Derived Heart Repair" Journal of Developmental Biology 2, no. 2: 84-100. https://doi.org/10.3390/jdb2020084
APA StyleSmits, A. M., & Riley, P. R. (2014). Epicardium-Derived Heart Repair. Journal of Developmental Biology, 2(2), 84-100. https://doi.org/10.3390/jdb2020084