Epicardial Origin of Resident Mesenchymal Stem Cells in the Adult Mammalian Heart

{kind=link}
{kind=link}
Abstract
:1. Overview of Epicardial Development
2. Epicardial Plasticity in the Embryo
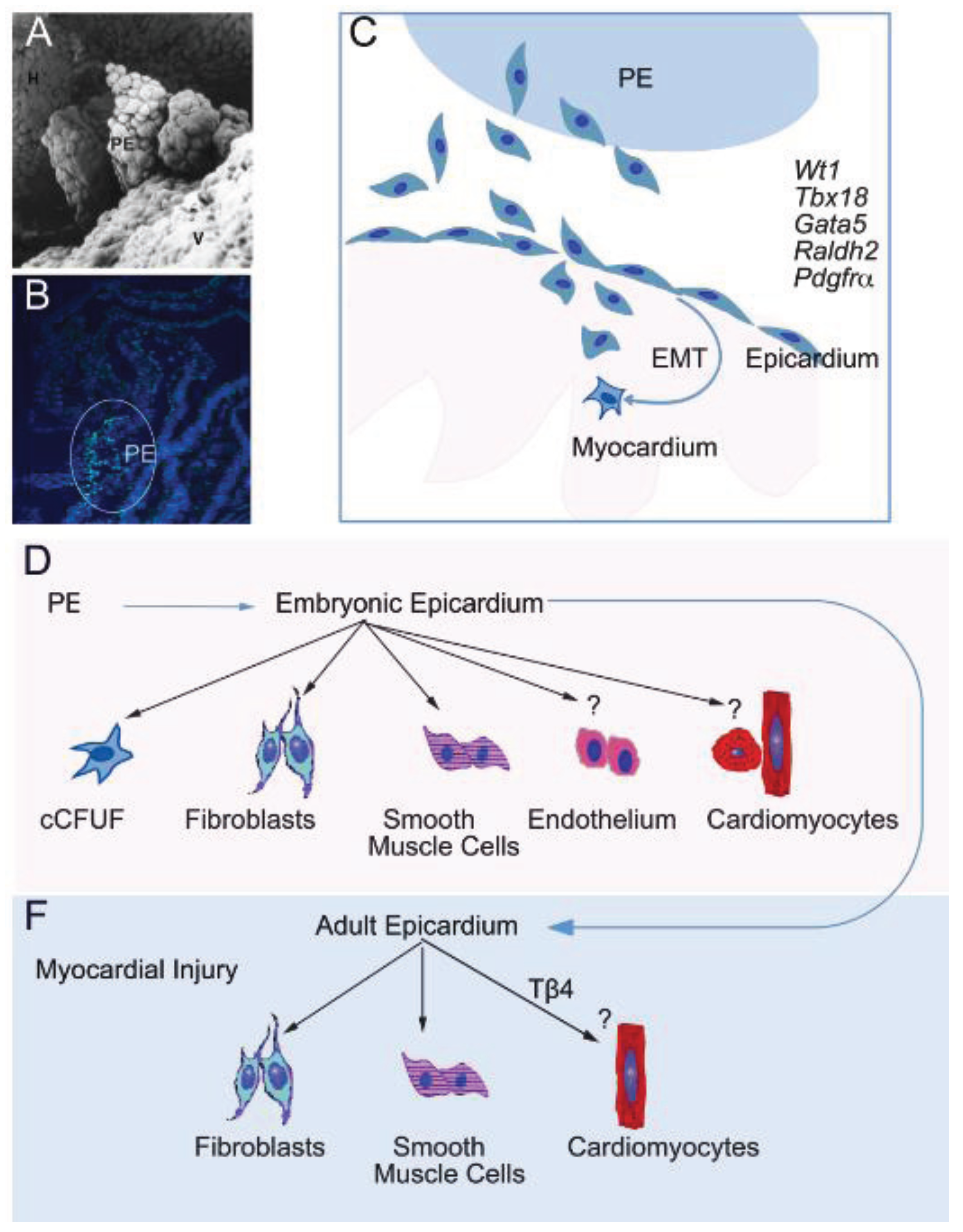
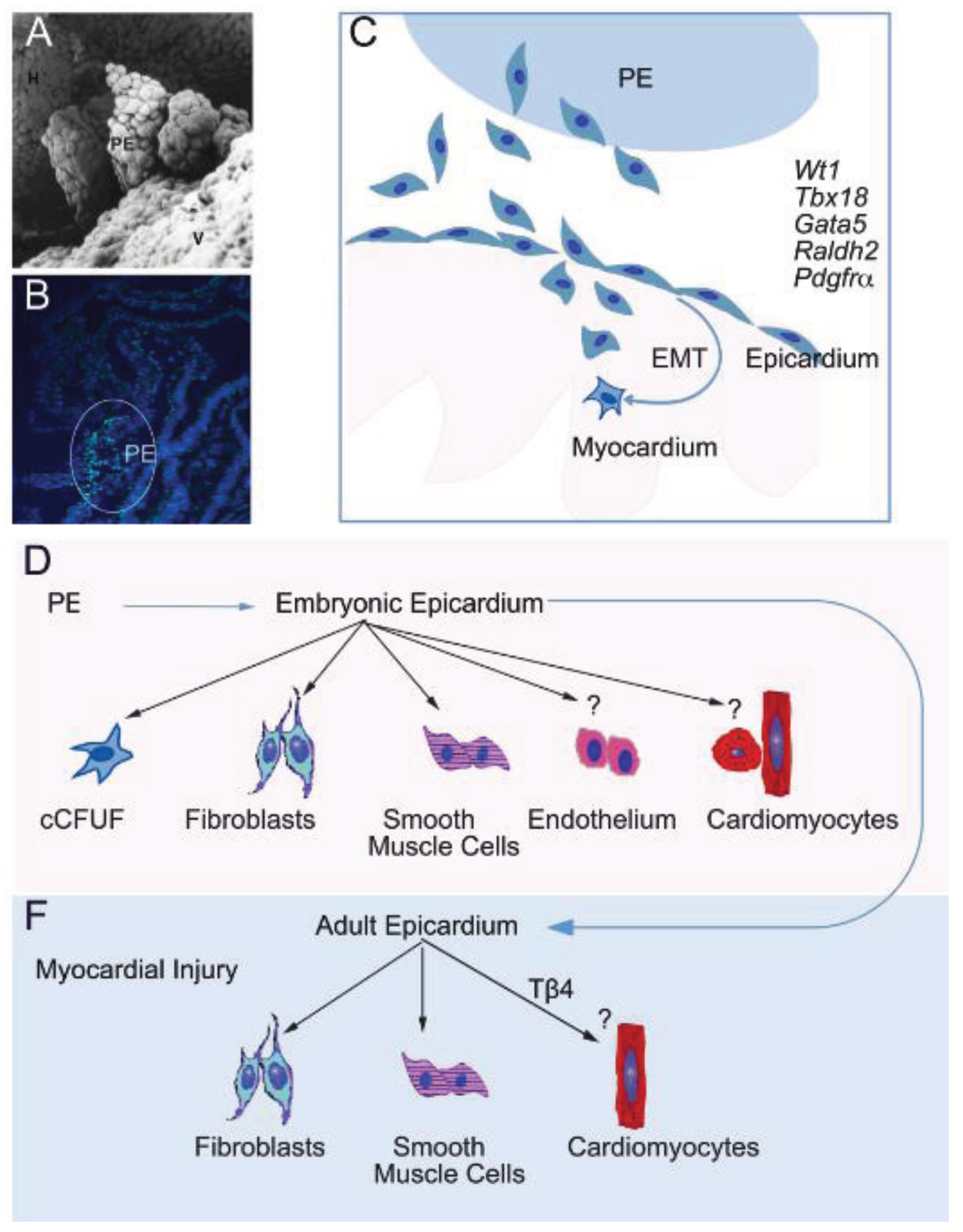
3. Adult Cardiac Stem/Progenitor Cells
4. Epicardium-Derived Stem/Progenitor Cells
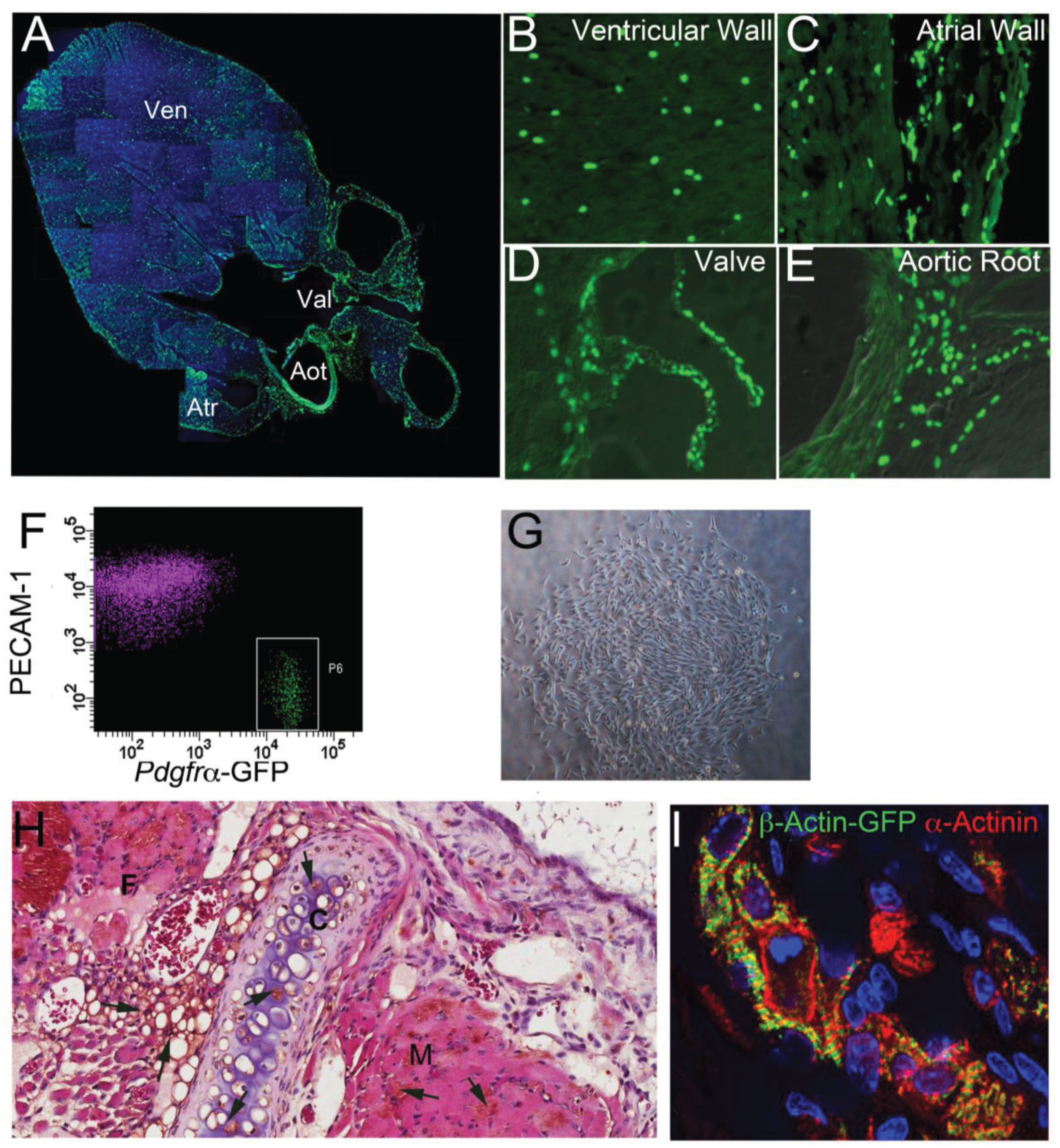
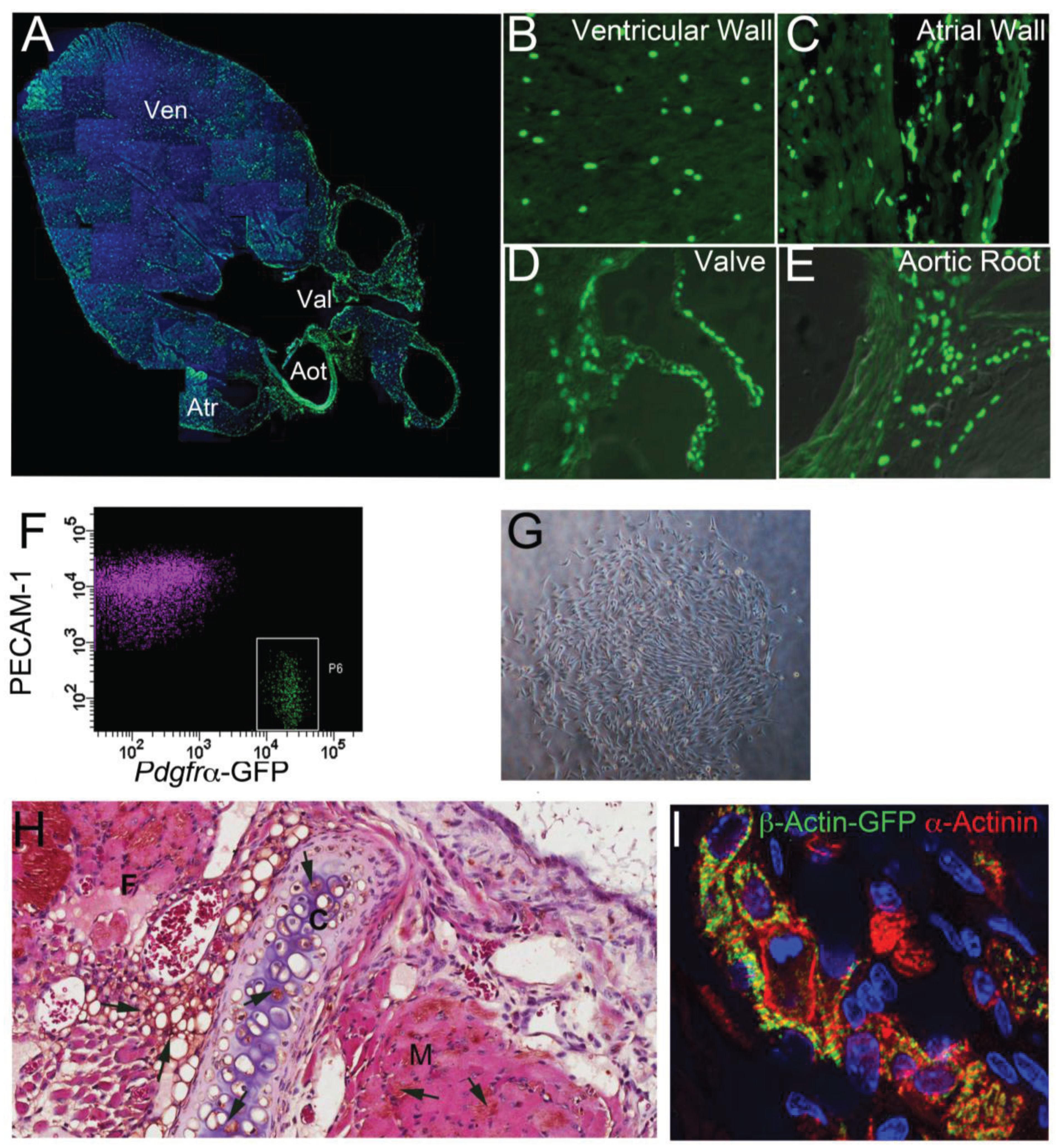
5. Lineage Potency of Cardiac MSC-Like Cells
6. The Adult Epicardium as a Regenerative Platform
7. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yung, S.; Chan, T.M. Intrinsic cells: Mesothelial cells—Central players in regulating inflammation and resolution. Perit Dial. Int. 2009, 29 (Suppl. 2), S21–S27. [Google Scholar]
- Rinkevich, Y.; Mori, T.; Sahoo, D.; Xu, P.X.; Bermingham, J.R., Jr.; Weissman, I.L. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat. Cell. Biol 2012, 14, 1251–1260. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. JCI 2011, 121, 1894–1904. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef]
- Bochmann, L.; Sarathchandra, P.; Mori, F.; Lara-Pezzi, E.; Lazzaro, D.; Rosenthal, N. Revealing new mouse epicardial cell markers through transcriptomics. PLoS ONE 2010, 5, e11429. [Google Scholar]
- Schlueter, J.; Brand, T. A right-sided pathway involving fgf8/snai1 controls asymmetric development of the proepicardium in the chick embryo. Proc. Natl. Acad. Sci. USA 2009, 106, 7485–7490. [Google Scholar] [CrossRef]
- Asli, N.S.; Harvey, R.P. Epithelial to mesenchymal transition as a portal to stem cell characters embedded in gene networks. BioEssays 2013, 35, 191–200. [Google Scholar] [CrossRef]
- Ishii, Y.; Langberg, J.D.; Hurtado, R.; Lee, S.; Mikawa, T. Induction of proepicardial marker gene expression by the liver bud. Development 2007, 134, 3627–3637. [Google Scholar] [CrossRef]
- Liu, J.; Stainier, D.Y. Tbx5 and bmp signaling are essential for proepicardium specification in zebrafish. Circ. Res. 2010, 106, 1818–1828. [Google Scholar] [CrossRef]
- Pombal, M.A.; Carmona, R.; Megias, M.; Ruiz, A.; Perez-Pomares, J.M.; Munoz-Chapuli, R. Epicardial development in lamprey supports an evolutionary origin of the vertebrate epicardium from an ancestral pronephric external glomerulus. Evol. Dev. 2008, 10, 210–216. [Google Scholar] [CrossRef]
- Zhou, B.; von Gise, A.; Ma, Q.; Rivera-Feliciano, J.; Pu, W.T. Nkx2–5- and isl1-expressing cardiac progenitors contribute to proepicardium. BBRC 2008, 375, 450–453. [Google Scholar]
- Kruithof, B.P.; van Wijk, B.; Somi, S.; Kruithof-de Julio, M.; Perez Pomares, J.M.; Weesie, F.; Wessels, A.; Moorman, A.F.; van den Hoff, M.J. Bmp and fgf regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev. Biol. 2006, 295, 507–522. [Google Scholar] [CrossRef]
- Schlueter, J.; Brand, T. A subpopulation of proepicardial cells is derived from the somatic mesoderm in the chick embryo. Circ. Res. 2013, 113, 1106–1108. [Google Scholar] [CrossRef]
- Schlueter, J.; Manner, J.; Brand, T. Bmp is an important regulator of proepicardial identity in the chick embryo. Dev. Biol. 2006, 295, 546–558. [Google Scholar] [CrossRef]
- Stennard, F.A.; Harvey, R.P. T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development 2005, 132, 4897–4910. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9; discussion E9–E10. [Google Scholar] [CrossRef]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An nkx2–5/bmp2/smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef]
- Singh, R.; Harvey, R.P. Proepicadrium is enlarged and hyperactive in nkx2–5 hypomorphic mutant mice. Unpublished work. 2014. [Google Scholar]
- Watt, A.J.; Battle, M.A.; Li, J.; Duncan, S.A. Gata4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc. Natl. Acad. Sci USA 2004, 101, 12573–12578. [Google Scholar] [CrossRef]
- Greulich, F.; Farin, H.F.; Schuster-Gossler, K.; Kispert, A. Tbx18 function in epicardial development. Cardiovasc. Res. 2012, 96, 476–483. [Google Scholar] [CrossRef]
- Wu, S.P.; Dong, X.R.; Regan, J.N.; Su, C.; Majesky, M.W. Tbx18 regulates development of the epicardium and coronary vessels. Dev. Biol. 2013, 383, 307–320. [Google Scholar] [CrossRef]
- Mikawa, T.; Gourdie, R.G. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 1996, 174, 221–232. [Google Scholar] [CrossRef]
- Ishii, Y.; Garriock, R.J.; Navetta, A.M.; Coughlin, L.E.; Mikawa, T. Bmp signals promote proepicardial protrusion necessary for recruitment of coronary vessel and epicardial progenitors to the heart. Dev. Cell. 2010, 19, 307–316. [Google Scholar] [CrossRef]
- Hatcher, C.J.; Diman, N.Y.; Kim, M.S.; Pennisi, D.; Song, Y.; Goldstein, M.M.; Mikawa, T.; Basson, C.T. A role for tbx5 in proepicardial cell migration during cardiogenesis. Physiol. Genomics 2004, 18, 129–140. [Google Scholar] [CrossRef]
- Munoz-Chapuli, R.; Macias, D.; Gonzalez-Iriarte, M.; Carmona, R.; Atencia, G.; Perez-Pomares, J.M. The epicardium and epicardial-derived cells: Multiple functions in cardiac development. Rev. Eep. Cardiol. 2002, 55, 1070–1082. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Bergwerff, M.; Mentink, M.M.; Poelmann, R.E. Epicardial outgrowth inhibition leads to compensatory mesothelial outflow tract collar and abnormal cardiac septation and coronary formation. Circ. Res. 2000, 87, 969–971. [Google Scholar] [CrossRef]
- Lie-Venema, H.; Gittenberger-de Groot, A.C.; van Empel, L.J.; Boot, M.J.; Kerkdijk, H.; de Kant, E.; DeRuiter, M.C. Ets-1 and ets-2 transcription factors are essential for normal coronary and myocardial development in chicken embryos. Circ. Res. 2003, 92, 749–756. [Google Scholar] [CrossRef]
- Wessels, A.; Perez-Pomares, J.M. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. 2004, 276, 43–57. [Google Scholar] [CrossRef]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of snail and e-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef]
- Carmona, R.; Guadix, J.A.; Cano, E.; Ruiz-Villalba, A.; Portillo-Sanchez, V.; Perez-Pomares, J.M.; Munoz-Chapuli, R. The embryonic epicardium: An essential element of cardiac development. J. Cell. Mol. Med. 2010, 14, 2066–2072. [Google Scholar] [CrossRef]
- Vrancken Peeters, M.P.; Gittenberger-de Groot, A.C.; Mentink, M.M.; Poelmann, R.E. Smooth muscle cells and fibroblasts of the coronary arteries derive from epithelial-mesenchymal transformation of the epicardium. Anat. Embryol. (Berl.) 1999, 199, 367–378. [Google Scholar] [CrossRef]
- Mikawa, T.; Borisov, A.; Brown, A.M.; Fischman, D.A. Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus: I. Formation of the ventricular myocardium. Dev. Dyn. 1992, 193, 11–23. [Google Scholar] [CrossRef]
- Red-Horse, K.; Ueno, H.; Weissman, I.L.; Krasnow, M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, Z.; Lui, W.; Chen, X.; Wang, Y.; Chamberlain, A.A.; Moreno-Rodriguez, R.A.; Markwald, R.R.; O’Rourke, B.P.; Sharp, D.J.; et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 2012, 151, 1083–1096. [Google Scholar] [CrossRef]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The BHLH transcription factor tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Merki, E.; Zamora, M.; Raya, A.; Kawakami, Y.; Wang, J.; Zhang, X.; Burch, J.; Kubalak, S.W.; Kaliman, P.; Izpisua Belmonte, J.C.; et al. Epicardial retinoid x receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 2005, 102, 18455–18460. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell. 2012, 22, 639–650. [Google Scholar]
- Rudat, C.; Kispert, A. Wt1 and epicardial fate mapping. Circ. Res. 2012, 111, 165–169. [Google Scholar] [CrossRef]
- Zhou, B.; Pu, W.T. Genetic Cre-loxp assessment of epicardial cell fate using wt1-driven cre alleles. Circ. Res. 2012, 111, e276–e280. [Google Scholar] [CrossRef]
- Buckingham, M.E.; Meilhac, S.M. Tracing cells for tracking cell lineage and clonal behavior. Dev. Cell. 2011, 21, 394–409. [Google Scholar] [CrossRef]
- Zeng, B.; Ren, X.F.; Cao, F.; Zhou, X.Y.; Zhang, J. Developmental patterns and characteristics of epicardial cell markers tbx18 and wt1 in murine embryonic heart. J. Biomed. Sci. 2011, 18. [Google Scholar] [CrossRef]
- Manner, J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anat. Rec. 1999, 255, 212–226. [Google Scholar] [CrossRef]
- van Wijk, B.; van den Berg, G.; Abu-Issa, R.; Barnett, P.; van der Velden, S.; Schmidt, M.; Ruijter, J.M.; Kirby, M.L.; Moorman, A.F.; van den Hoff, M.J. Epicardium and myocardium separate from a common precursor pool by crosstalk between bone morphogenetic protein- and fibroblast growth factor-signaling pathways. Circ. Res. 2009, 105, 431–441. [Google Scholar] [CrossRef]
- Bock-Marquette, I.; Shrivastava, S.; Pipes, G.C.; Thatcher, J.E.; Blystone, A.; Shelton, J.M.; Galindo, C.L.; Melegh, B.; Srivastava, D.; Olson, E.N.; et al. Thymosin beta4 mediated pkc activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J. Mol. Cell. Cardiol. 2009, 46, 728–738. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Schedl, A. The complex life of wt1. JCS 2003, 116, 1653–1658. [Google Scholar] [CrossRef]
- Chong, J.J.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult cardiac-resident msc-like stem cells with a proepicardial origin. Cell. Stem Cell. 2011, 9, 527–540. [Google Scholar] [CrossRef]
- Grieskamp, T.; Rudat, C.; Ludtke, T.H.; Norden, J.; Kispert, A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ. Res. 2011, 108, 813–823. [Google Scholar] [CrossRef]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. Tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef]
- Gonzalez-Rosa, J.M.; Peralta, M.; Mercader, N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev. Biol. 2012, 370, 173–186. [Google Scholar] [CrossRef]
- Mikawa, T.; Fischman, D.A. Retroviral analysis of cardiac morphogenesis: Discontinuous formation of coronary vessels. Proc. Natl. Acad. Sci. USA 1992, 89, 9504–9508. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Laugwitz, K.L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.Z.; Cai, C.L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef]
- Martin, C.M.; Meeson, A.P.; Robertson, S.M.; Hawke, T.J.; Richardson, J.A.; Bates, S.; Goetsch, S.C.; Gallardo, T.D.; Garry, D.J. Persistent expression of the atp-binding cassette transporter, abcg2, identifies cardiac sp cells in the developing and adult heart. Dev. Biol. 2004, 265, 262–275. [Google Scholar] [CrossRef]
- Meinhardt, A.; Spicher, A.; Roehrich, M.E.; Glauche, I.; Vogt, P.; Vassalli, G. Immunohistochemical and flow cytometric analysis of long-term label-retaining cells in the adult heart. Stem Cells Dev. 2011, 20, 211–222. [Google Scholar] [CrossRef]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marban, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef]
- Bolli, R.; Chugh, A.R.; D'Amario, D.; Loughran, J.H.; Stoddard, M.F.; Ikram, S.; Beache, G.M.; Wagner, S.G.; Leri, A.; Hosoda, T.; et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): Initial results of a randomised phase 1 trial. Lancet 2011, 378, 1847–1857. [Google Scholar] [CrossRef]
- Fazel, S.; Cimini, M.; Chen, L.; Li, S.; Angoulvant, D.; Fedak, P.; Verma, S.; Weisel, R.D.; Keating, A.; Li, R.K. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. JCI 2006, 116, 1865–1877. [Google Scholar] [CrossRef]
- Ferreira-Martins, J.; Ogorek, B.; Cappetta, D.; Matsuda, A.; Signore, S.; D’Amario, D.; Kostyla, J.; Steadman, E.; Ide-Iwata, N.; Sanada, F.; et al. Cardiomyogenesis in the developing heart is regulated by c-kit-positive cardiac stem cells. Circ. Res. 2012, 110, 701–715. [Google Scholar] [CrossRef]
- Wu, S.M.; Fujiwara, Y.; Cibulsky, S.M.; Clapham, D.E.; Lien, C.L.; Schultheiss, T.M.; Orkin, S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 2006, 127, 1137–1150. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell. Tissue Kinet 1970, 3, 393–403. [Google Scholar]
- Pelekanos, R.A.; Li, J.; Gongora, M.; Chandrakanthan, V.; Scown, J.; Suhaimi, N.; Brooke, G.; Christensen, M.E.; Doan, T.; Rice, A.M.; et al. Comprehensive transcriptome and immunophenotype analysis of renal and cardiac MSC-like populations supports strong congruence with bone marrow MSC despite maintenance of distinct identities. Stem Cell. Res. 2012, 8, 58–73. [Google Scholar] [CrossRef]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef]
- Chan, C.K.; Lindau, P.; Jiang, W.; Chen, J.Y.; Zhang, L.F.; Chen, C.C.; Seita, J.; Sahoo, D.; Kim, J.B.; Lee, A.; et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12643–12648. [Google Scholar] [CrossRef]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef]
- Tavassoli, M.; Crosby, W.H. Transplantation of marrow to extramedullary sites. Science 1968, 161, 54–56. [Google Scholar]
- Hamilton, T.G.; Klinghoffer, R.A.; Corrin, P.D.; Soriano, P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. J. Mol. Cell. Biol. 2003, 23, 4013–4025. [Google Scholar] [CrossRef]
- Reinhard, H.; Harvey, R.P. Cardiac pdgfra-positive cells lie outside of the endothelial basement membrane. Unpublished work. 2014. [Google Scholar]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–26. [Google Scholar] [CrossRef]
- Prall, O.J.W.; Chandrakanthan, V.; Harvey, R.P. Colony-forming activity is diminished in pdgfr-alpha hypomorphic fetuses. Unpublished work. 2014. [Google Scholar]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell. Stem Cell. 2013, 13, 392–402. [Google Scholar] [CrossRef]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.T.; Shaw, R.M.; Srivastava, D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell. 2009, 16, 233–244. [Google Scholar] [CrossRef]
- Judson, R.N.; Zhang, R.H.; Rossi, F.M. Tissue-resident mesenchymal stem/progenitor cells in skeletal muscle: Collaborators or saboteurs? FEBS J. 2013, 280, 4100–4108. [Google Scholar] [CrossRef]
- Liu, X.; Tan, M.; Gong, D.; Han, L.; Lu, F.; Huang, S.; Xu, Z. Characteristics of pericardial interstitial cells and their implications in pericardial fibrocalcification. J. Mol. Cell. Cardiol. 2012, 53, 780–789. [Google Scholar] [CrossRef]
- Nam, Y.J.; Song, K.; Olson, E.N. Heart repair by cardiac reprogramming. Nat. Med. 2013, 19, 413–415. [Google Scholar] [CrossRef]
- Wills, A.A.; Holdway, J.E.; Major, R.J.; Poss, K.D. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development 2008, 135, 183–192. [Google Scholar]
- Kocabas, F.; Mahmoud, A.I.; Sosic, D.; Porrello, E.R.; Chen, R.; Garcia, J.A.; DeBerardinis, R.J.; Sadek, H.A. The hypoxic epicardial and subepicardial microenvironment. Cardiovasc. Transl. Res. 2012, 5, 654–665. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Popescu, L.M. Cardiomyocyte precursors and telocytes in epicardial stem cell niche: Electron microscope images. J. Cell. Mol. Med. 2010, 14, 871–877. [Google Scholar] [CrossRef]
- Bax, N.A.; Pijnappels, D.A.; van Oorschot, A.A.; Winter, E.M.; de Vries, A.A.; van Tuyn, J.; Braun, J.; Maas, S.; Schalij, M.J.; Atsma, D.E.; et al. Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J. Cell. Mol. Med. 2011, 15, 2675–2683. [Google Scholar] [CrossRef]
- van Tuyn, J.; Atsma, D.E.; Winter, E.M.; van der Velde-van Dijke, I.; Pijnappels, D.A.; Bax, N.A.; Knaan-Shanzer, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; van der Laarse, A.; et al. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells 2007, 25, 271–278. [Google Scholar] [CrossRef]
- Di Meglio, F.; Castaldo, C.; Nurzynska, D.; Miraglia, R.; Romano, V.; Russolillo, V.; Giuseppina, L.; Vosa, C.; Montagnani, S. Localization and origin of cardiac cd117-positive cells: Identification of a population of epicardially-derived cells in adult human heart. Ital. J. Anat Embryol. 2010, 115, 71–78. [Google Scholar]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Poelmann, R.E. Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Mol. Cell. Med. 2010, 14, 1056–1060. [Google Scholar]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De Mori, R.; Marchetti, C.; Pozzoli, O.; et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit(+) cells: Role of the pericardial fluid. J. Mol. Cell. Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Melville, A.A.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; Ma, Q.; Oh, J.H.; Lin, R.Z.; Melero-Martin, J.M.; von Gise, A.; Zhou, P.; Hu, T.; He, L.; et al. Thymosin beta 4 treatment after myocardial infarction does not reprogram epicardial cells into cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 43–47. [Google Scholar] [CrossRef]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Asli, N.S.; Xaymardan, M.; Harvey, R.P. Epicardial Origin of Resident Mesenchymal Stem Cells in the Adult Mammalian Heart. J. Dev. Biol. 2014, 2, 117-137. https://doi.org/10.3390/jdb2020117
Asli NS, Xaymardan M, Harvey RP. Epicardial Origin of Resident Mesenchymal Stem Cells in the Adult Mammalian Heart. Journal of Developmental Biology. 2014; 2(2):117-137. https://doi.org/10.3390/jdb2020117
Chicago/Turabian StyleAsli, Naisana S., Munira Xaymardan, and Richard P. Harvey. 2014. "Epicardial Origin of Resident Mesenchymal Stem Cells in the Adult Mammalian Heart" Journal of Developmental Biology 2, no. 2: 117-137. https://doi.org/10.3390/jdb2020117
APA StyleAsli, N. S., Xaymardan, M., & Harvey, R. P. (2014). Epicardial Origin of Resident Mesenchymal Stem Cells in the Adult Mammalian Heart. Journal of Developmental Biology, 2(2), 117-137. https://doi.org/10.3390/jdb2020117