

Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies


Abstract
1. Introduction
2. Primary Cilia Deficits in CNS Disorders: Ciliopathies and Beyond
3. Primary Cilia Defects in Monogenic Neurodevelopmental Disorders
3.1. Fragile X Syndrome
3.2. Tuberous Sclerosis Complex
3.3. Focal Cortical Dysplasia
3.4. Cyclin-Dependent Kinase-like 5 Deficiency Disorder
3.5. Rett Syndrome
4. Ciliary Defects as a Convergent Mechanism for Neurologic Phenotypes
Crosstalk between Signaling Pathways in Monogenic Neurodevelopmental Disorders
5. Impact of Primary Cilia Loss in Neurons and Neuronal Networks
Primary Cilia Signaling Pathways in Brain Pathology
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC3 | Type 3 adenyl cyclase |
| ARF | ADP-ribosylation factors |
| ASD | Autism spectrum disorder |
| CDD | CDKL5 deficiency disorder |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CNS | Central nervous system |
| DG | Dentate gyrus |
| DGC | Dentate granule cell |
| DNe | Dentate neuroepithelium |
| E/I | Excitation/inhibition |
| FCD | Focal cortical dysplasia |
| FGF | Fibroblast growth factor |
| FMCD | Focal malformations of cortical development |
| FMRP | Fragile X messenger ribonucleoprotein |
| FXS | Fragile X syndrome |
| GluD1 | Glutamate D1 receptor |
| GPCR | G-protein-coupled receptor |
| HDAC6 | Histone deacetylase 6 |
| Hsp27 | Heat shock protein 27 |
| ID | Intellectual disability |
| IFT | Intraflagellar transport |
| KD | Knockdown |
| KO | Knockout |
| LTP | Long term potentiation |
| MCHR1 | Melanin concentrating hormone receptor 1 |
| MeCP2 | Methyl-CpG-binding protein 2 |
| MEF | Mouse embryonic fibroblast |
| mTOR | Mechanistic target of rapamycin |
| OFD1 | Oral-facial-digital syndrome 1 protein |
| PDGFα | Platelet-derived growth factor α |
| PHTS | PTEN hamartoma tumor syndrome |
| PKD | Polycystic kidney disease |
| p-rpS6 | Phosphorylated ribosomal protein S6 |
| PTEN | Phosphatase and tensin homolog |
| RTT | Rett syndrome |
| Shh | Sonic hedgehog |
| SGZ | Subgranular zone |
| SSTR3 | Somatostatin receptor type 3 |
| TEM | Transmission electron microscopy |
| TGF-β | Transforming growth factor β |
| TSC | Tuberous sclerosis complex |
| Ttbk2 | Tau tubulin kinase 2 |
| 5-HT | 5-hydroxytryptamine |
| 5-HTR6 | 5-HT receptor type 6 |
References
- Leeuwenhoek, A. Concerning little animals observed in rain-, well-, sea- and snow-water; as also in water wherein pepper had lain infused. Philos. Trans. 1677, 12, 821–831. [Google Scholar]
- Kiesel, P.; Viar, G.A.; Tsoy, N.; Maraspini, R.; Gorilak, P.; Varga, V.; Honigmann, A.; Pigino, G. The molecular structure of mammalian primary cilia revealed by cryo-electron tomography. Nat. Struct. Mol. Biol. 2020, 27, 1115–1124. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Bloodgood, R.A. From Central to Rudimentary to Primary: The History of an Underappreciated Organelle Whose Time Has Come. The Primary Cilium. Methods Cell Biol. 2009, 94, 2–52. [Google Scholar]
- Zimmermann, K.W. Beiträge zur Kenntniss einiger Drüsen und Epithelien. Arch. Für Mikrosk. Anat. 1898, 52, 552–706. [Google Scholar] [CrossRef]
- Sjostrand, F.S. The ultrastructure of the innersegments of the retinal rods of the guinea pig eye as revealed by electron microscopy. J. Cell Comp. Physiol. 1953, 42, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, D.W.; Porter, K.R. A study of the fine structure of ciliated epithelia. J. Morphol. 1954, 94, 221–281. [Google Scholar] [CrossRef]
- De Robertis, E. Electron microscope observations on the submicroscopic organization of the retinal rods. J. Biophys. Biochem. Cytol. 1956, 2, 319–330. [Google Scholar] [CrossRef]
- Sorokin, S. Centrioles and The Formation of Rudimentary Cilia by Fibroblasts and Smooth Muscle Cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef]
- Flood, P.R.; Totland, G.K. Substructure of solitary cilia in mouse kidney. Cell Tissue Res. 1977, 183, 281–290. [Google Scholar] [CrossRef]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Izawa, I.; Goto, H.; Kasahara, K.; Inagaki, M. Current topics of functional links between primary cilia and cell cycle. Cilia 2015, 4, 1–13. [Google Scholar] [CrossRef]
- Smith, C.E.L.; Lake, A.V.R.; Johnson, C.A. Primary Cilia, Ciliogenesis and the Actin Cytoskeleton: A Little Less Resorption, A Little More Actin Please. Front. Cell Dev. Biol. 2020, 8, 622822. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef]
- Klena, N.; Pigino, G. Structural Biology of Cilia and Intraflagellar Transport. Annu. Rev. Cell Dev. Biol. 2022, 38, 103–123. [Google Scholar] [CrossRef]
- Wood, C.R.; Rosenbaum, J.L. Ciliary ectosomes: Transmissions from the cell’s antenna. Trends Cell Biol. 2015, 25, 276–285. [Google Scholar] [CrossRef]
- Long, H.; Huang, K. Transport of Ciliary Membrane Proteins. Front. Cell Dev. Biol. 2019, 7, 381. [Google Scholar] [CrossRef]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Del Cerro, M.P.; Snider, R.S. The Purkinje cell cilium. Anat. Rec. 1969, 165, 127–130. [Google Scholar] [CrossRef]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef]
- Spassky, N.; Han, Y.-G.; Aguilar, A.; Strehl, L.; Besse, L.; Laclef, C.; Ros, M.R.; Garcia-Verdugo, J.; Alvarez-Buylla, A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 2008, 317, 246–259. [Google Scholar] [CrossRef]
- Han, Y.-G.; Spassky, N.; Romaguera-Ros, M.; Garcia-Verdugo, J.-M.; Aguilar, A.; Schneider-Maunoury, S.; Alvarez-Buylla, A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 2008, 11, 277–284. [Google Scholar] [CrossRef]
- Sipos, É.; Komoly, S.; Ács, P. Quantitative Comparison of Primary Cilia Marker Expression and Length in the Mouse Brain. J. Mol. Neurosci. 2018, 64, 397–409. [Google Scholar] [CrossRef]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu. Rev. Genom. Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Lee, J.E.; Gleeson, J.G. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011, 3, 59. [Google Scholar] [CrossRef]
- Louvi, A.; Grove, E.A. Cilia in the CNS: The Quiet Organelle Claims Center Stage. Neuron 2011, 69, 1046–1060. [Google Scholar] [CrossRef]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef]
- Muñoz-Estrada, J.; Lora-Castellanos, A.; Meza, I.; Elizalde, S.A.; Benítez-King, G. Primary cilia formation is diminished in schizophrenia and bipolar disorder: A possible marker for these psychiatric diseases. Schizophr. Res. 2018, 195, 412–420. [Google Scholar] [CrossRef]
- Ma, R.; Kutchy, N.A.; Chen, L.; Meigs, D.D.; Hu, G. Primary cilia and ciliary signaling pathways in aging and age-related brain disorders. Neurobiol. Dis. 2022, 163, 105607. [Google Scholar] [CrossRef]
- Ki, S.M.; Jeong, H.S.; Lee, J.E. Primary Cilia in Glial Cells: An Oasis in the Journey to Overcoming Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 736888. [Google Scholar] [CrossRef]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.-M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J. Psychiatr. Res. 2018, 107, 138–144. [Google Scholar] [CrossRef]
- Lee, B.; Panda, S.; Lee, H.Y. Primary Ciliary Deficits in the Dentate Gyrus of Fragile X Syndrome. Stem Cell Rep. 2020, 15, 454–466. [Google Scholar] [CrossRef]
- Razak, K.A.; Dominick, K.C.; Erickson, C.A. Developmental studies in fragile X syndrome. J. Neurodev. Disord. 2020, 12, 13–15. [Google Scholar] [CrossRef]
- Di Nardo, A.; Lenoel, I.; Winden, K.D.; Ruhmkorf, A.; Modi, M.E.; Barrett, L.; Marzelli, A. Phenotypic Screen with TSC-Deficient Neurons Reveals Heat-Shock Machinery as a Druggable Pathway for mTORC1 and Reduced Cilia. Cell Rep. 2020, 31, 107780. [Google Scholar] [CrossRef]
- Crino, P.B. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013, 125, 317–332. [Google Scholar] [CrossRef]
- Di Mario, F.J., Jr.; Sahin, M.; Ebrahimi-Fakhari, D. Tuberous sclerosis complex. Pediatr. Clin. N. Am. 2015, 62, 633–648. [Google Scholar] [CrossRef]
- Kabat, J.; Krol, P. Focal cortical dysplasia-review. Pol. J. Radiol. 2012, 77, 35–43. [Google Scholar] [CrossRef]
- Lim, K.-C.; Crino, P. Focal malformations of cortical development: New vistas for molecular pathogenesis. Neuroscience 2013, 252, 262–276. [Google Scholar] [CrossRef]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7. [Google Scholar] [CrossRef]
- Di Nardo, A.; Ruhmkorf, A.; Award, P.; Brennecke, A.; Fagiolini, M.; Sahin, M. Phenotypic characterization of Cdkl5-knockdown neurons establishes elongated cilia as a functional assay for CDKL5 Deficiency Disorder. Neurosci. Res. 2022, 176, 73–78. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131 Pt 10, 2647–2661. [Google Scholar] [CrossRef]
- Frasca, A.; Spiombi, E.; Palmieri, M.; Albizzati, E.; Valente, M.M.; Bergo, A.; Leva, B.; Kilstrup-Nielsen, C.; Bianchi, F.; Di Carlo, V.; et al. MECP2 mutations affect ciliogenesis: A novel perspective for Rett syndrome and related disorders. EMBO Mol. Med. 2020, 12, e10270. [Google Scholar] [CrossRef]
- Smeets, E.E.; Pelc, K.; Dan, B. Rett Syndrome. Mol. Syndromol. 2012, 2, 113–127. [Google Scholar] [CrossRef]
- Yu, S.; Pritchard, M.; Kremer, E.; Lynch, M.; Nancarrow, J.; Baker, E.; Holman, K.; Mulley, J.C.; Warren, S.T.; Schlessinger, D.; et al. Fragile X Genotype Characterized by an Unstable Region of DNA. Science 1991, 252, 1179–1181. [Google Scholar] [CrossRef]
- Lee, B.; Beuhler, L.; Lee, H.Y. The Primary Ciliary Deficits in Cerebellar Bergmann Glia of the Mouse Model of Fragile X Syndrome. Cerebellum 2022, 21, 801–813. [Google Scholar] [CrossRef]
- Uysal, S.P.; Şahin, M. Tuberous sclerosis: A review of the past, present, and future. Turk. J. Med Sci. 2020, 50, 1665–1676. [Google Scholar] [CrossRef]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef]
- Rosengren, T.; Larsen, L.J.; Pedersen, L.B.; Christensen, S.T.; Møller, L.B. TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell. Mol. Life Sci. 2018, 75, 2663–2680. [Google Scholar] [CrossRef]
- Armour, E.A.; Carson, R.P.; Ess, K.C. Cystogenesis and elongated primary cilia in Tsc1-deficient distal convoluted tubules. Am. J. Physiol. Physiol. 2012, 303, F584–F592. [Google Scholar] [CrossRef]
- Krueger, D.A.; Sadhwani, A.; Byars, A.W.; De Vries, P.J.; Franz, D.N.; Whittemore, V.H.; Filip-Dhima, R.; Murray, D.; Kapur, K.; Sahin, M. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 2017, 4, 877–887. [Google Scholar] [CrossRef]
- Overwater, I.E.; Rietman, A.B.; Mous, S.E.; Heus, K.B.-D.; Rizopoulos, D.; Hoopen, L.W.T.; Van Der Vaart, T.; Jansen, F.E.; Elgersma, Y.; Moll, H.A.; et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 2019, 93, e200–e209. [Google Scholar] [CrossRef]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef]
- Shao, D.D.; Achkar, C.M.; Lai, A.; Srivastava, S.; Doan, R.N.; Rodan, L.H.; Chen, A.Y.; Poduri, A.; Yang, E.; Walsh, C.A.; et al. Polymicrogyria is Associated With Pathogenic Variants in PTEN. Ann. Neurol. 2020, 88, 1153–1164. [Google Scholar] [CrossRef]
- Nabbout, R.; Dulac, O. Epilepsy. Genetics of early-onset epilepsy with encephalopathy. Nat. Rev. Neurol. 2012, 8, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Shaw, C.A.; Dawson, B.C.; Dugas, D.V.; Al-Mohtaseb, Z.; Hill, D.E.; Zoghbi, H.Y. Protein Interactome Reveals Converging Molecular Pathways Among Autism Disorders. Sci. Transl. Med. 2011, 3, 86ra49. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing Chromosomal Abnormalities Reveals Neurodevelopmental Loci that Confer Risk across Diagnostic Boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 Expression Is Modulated during Neuronal Development and Its Subcellular Distribution Is Tightly Regulated by the C-terminal Tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.-W.; Ranum, P.T.; Lefebvre, P.A. CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Mol. Biol. Cell 2013, 24, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Valente, D.; Chandola, C.; Magi, F.; Bergo, A.; Monteonofrio, L.; Tramarin, M.; Fazzari, M.; Soddu, S.; Landsberger, N.; et al. CDKL5 localizes at the centrosome and midbody and is required for faithful cell division. Sci. Rep. 2017, 7, 6228. [Google Scholar] [CrossRef]
- Canning, P.; Park, K.; Gonçalves, J.; Li, C.; Howard, C.; Sharpe, T.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Banerjee, A.; Castro, J.; Sur, M. Rett syndrome: Genes, synapses, circuits, and therapeutics. Front. Psychiatry 2012, 3, 34. [Google Scholar] [CrossRef]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; LaSalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzo, A.; De Felice, C.; Guy, J.; Timperio, A.M.; Zolla, L.; Guerranti, R.; Leoncini, S.; Signorini, C.; Durand, T.; Hayek, J. Brain protein changes in Mecp2 mouse mutant models: Effects on disease progression of Mecp2 brain specific gene reactivation. J. Proteom. 2019, 210, 103537. [Google Scholar] [CrossRef]
- Bergo, A.; Strollo, M.; Gai, M.; Barbiero, I.; Stefanelli, G.; Sertic, S.; Bergo, M. Methyl-CpG binding protein 2 (MeCP2) localizes at the centrosome and is required for proper mitotic spindle organization. J. Biol. Chem. 2015, 290, 3223–3237. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Mariappan, R.; De, K.; Ohn, T. Loss of MeCP2 causes subtle alteration in dendritic arborization of retinal ganglion cells. Anim. Cells Syst. 2021, 25, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716. [Google Scholar] [CrossRef]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Vallero, A. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef]
- Rangasamy, S.; Olfers, S.; Gerald, B.; Hilbert, A.; Svejda, S.; Narayanan, V. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Research 2016, 5, 2269. [Google Scholar] [CrossRef]
- Ricciardi, S.; Boggio, E.M.; Grosso, S.; Lonetti, G.; Forlani, G.; Stefanelli, G.; Calcagno, E.; Morello, N.; Landsberger, N.; Biffo, S.; et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 2011, 20, 1182–1196. [Google Scholar] [CrossRef]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR Signaling in Fragile X Syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Arsenault, J.; Hooper, A.W.M.; Gholizadeh, S.; Kong, T.; Pacey, L.K.; Koxhioni, E.; Niibori, Y.; Eubanks, J.H.; Wang, L.-Y.; Hampson, D.R. Interregulation between fragile X mental retardation protein and methyl CpG binding protein 2 in the mouse posterior cerebral cortex. Hum. Mol. Genet. 2020, 29, 3744–3756. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.; Yuan, L.; Seong, E.; Ligon, C.; DeKorver, N.; Gurumurthy, C.; Arikkath, J. Neuron-Type Specific Loss of CDKL5 Leads to Alterations in mTOR Signaling and Synaptic Markers. Mol. Neurobiol. 2018, 56, 4151–4162. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef]
- Lai, Y.; Jiang, Y. Reciprocal Regulation between Primary Cilia and mTORC1. Genes 2020, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Gödel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef]
- Zhong, M.; Zhao, X.; Li, J.; Yuan, W.; Yan, G.; Tong, M.; Guo, S.; Zhu, Y.; Jiang, Y.; Liu, Y.; et al. Tumor Suppressor Folliculin Regulates mTORC1 through Primary Cilia. J. Biol. Chem. 2016, 291, 11689–11697. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Takahashi, K.; Nagai, T.; Chiba, S.; Nakayama, K.; Mizuno, K. Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell Sci. 2018, 131, jcs208769. [Google Scholar] [CrossRef]
- Sherpa, R.T.; Atkinson, K.F.; Ferreira, V.P.; Nauli, S.M. Rapamycin Increases Length and Mechanosensory Function Of Primary Cilia In Renal Epithelial And Vascular Endothelial Cells. IERJ 2016, 2, 91–97. [Google Scholar]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Larson, L. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef] [PubMed]
- Carouge, D.; Host, L.; Aunis, D.; Zwiller, J.; Anglard, P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol. Dis. 2010, 38, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Livide, G.; Patriarchi, T.; Amenduni, M.; Amabile, S.; Yasui, D.; Calcagno, E.; Rizzo, C.L.; De Falco, G.; Ulivieri, C.; Ariani, F.; et al. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet. 2015, 23, 195–201. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, H.J.; Lee, J.H. Roles of Primary Cilia in the Developing Brain. Front. Cell. Neurosci. 2019, 13, 218. [Google Scholar] [CrossRef]
- Hasenpusch-Theil, K.; Theil, T. The Multifaceted Roles of Primary Cilia in the Development of the Cerebral Cortex. Front. Cell Dev. Biol. 2021, 9, 630161. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Coufal, N.G.; Gleeson, J.G. Primary Cilia in the Developing and Mature Brain. Neuron 2014, 82, 511–521. [Google Scholar] [CrossRef]
- Suciu, S.K.; Caspary, T. Cilia, neural development and disease. Semin. Cell Dev. Biol. 2021, 110, 34–42. [Google Scholar] [CrossRef]
- Adamantidis, A.; Thomas, E.; Foidart, A.; Tyhon, A.; Coumans, B.; Minet, A.; Tirelli, E.; Seutin, V.; Grisar, T.; Lakaye, B. Disrupting the melanin-concentrating hormone receptor 1 in mice leads to cognitive deficits and alterations of NMDA receptor function. Eur. J. Neurosci. 2005, 21, 2837–2844. [Google Scholar] [CrossRef]
- May, S.R.; Ashique, A.M.; Karlen, M.; Wang, B.; Shen, Y.; Zarbalis, K.; Gleeson, M. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005, 287, 378–389. [Google Scholar] [CrossRef]
- Komada, M.; Saitsu, H.; Kinboshi, M.; Miura, T.; Shiota, K.; Ishibashi, M. Hedgehog signaling is involved in development of the neocortex. Development 2008, 135, 2717–2727. [Google Scholar] [CrossRef] [PubMed]
- Willaredt, M.A.; Hasenpusch-Theil, K.; Gardner, H.A.R.; Kitanovic, I.; Hirschfeld-Warneken, V.C.; Gojak, C.P.; Gorgas, K.; Bradford, C.L.; Spatz, J.; Wölfl, S.; et al. A Crucial Role for Primary Cilia in Cortical Morphogenesis. J. Neurosci. 2008, 28, 12887–12900. [Google Scholar] [CrossRef] [PubMed]
- Einstein, E.B.; Patterson, C.A.; Hon, B.J.; Regan, K.A.; Reddi, J.; Melnikoff, D.E.; Mariani, P. Somatostatin signaling in neuronal cilia is critical for object recognition memory. J. Neurosci. 2010, 30, 4306–4314. [Google Scholar] [CrossRef]
- Amador-Arjona, A.; Elliott, J.; Miller, A.; Ginbey, A.; Pazour, G.J.; Enikolopov, G.; Roberts, A.J.; Terskikh, A.V. Primary Cilia Regulate Proliferation of Amplifying Progenitors in Adult Hippocampus: Implications for Learning and Memory. J. Neurosci. 2011, 31, 9933–9944. [Google Scholar] [CrossRef]
- Besse, L.; Neti, M.; Anselme, I.; Gerhardt, C.; Rüther, U.; Laclef, C.; Schneider-Maunoury, S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 2011, 138, 2079–2088. [Google Scholar] [CrossRef]
- Wang, Z.; Phan, T.; Storm, D.R. The Type 3 Adenylyl Cyclase Is Required for Novel Object Learning and Extinction of Contextual Memory: Role of cAMP Signaling in Primary Cilia. J. Neurosci. 2011, 31, 5557–5561. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, H.; Eom, T.-Y.; Mariani, L.E.; Bachleda, A.; Hirt, J.; Gukassyan, V.; Cusack, C.L.; Lai, C.; Caspary, T.; Anton, E. Arl13b in Primary Cilia Regulates the Migration and Placement of Interneurons in the Developing Cerebral Cortex. Dev. Cell 2012, 23, 925–938. [Google Scholar] [CrossRef]
- Kumamoto, N.; Gu, Y.; Wang, J.; Janoschka, S.; Takemaru, K.-I.; Levine, J.; Ge, S. A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nat. Neurosci. 2012, 15, 399–405. [Google Scholar] [CrossRef]
- Higginbotham, H.; Guo, J.; Yokota, Y.; Umberger, N.L.; Su, C.-Y.; Li, J.; Verma, N.; Hirt, J.; Ghukasyan, V.; Caspary, T.; et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci. 2013, 16, 1000–1007. [Google Scholar] [CrossRef]
- Rhee, S.; Kirschen, G.W.; Gu, Y.; Ge, S. Depletion of primary cilia from mature dentate granule cells impairs hippocampus-dependent contextual memory. Sci. Rep. 2016, 6, 34370. [Google Scholar] [CrossRef]
- Gazea, M.; Tasouri, E.; Tolve, M.; Bosch, V.; Kabanova, A.; Gojak, C.; Kurtulmus, B.; Novikov, O.; Spatz, J.; Pereira, G.; et al. Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 2016, 409, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Foerster, P.; Daclin, M.; Asm, S.; Faucourt, M.; Boletta, A.; Genovesio, A.; Spassky, N. mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 2017, 144, 201–210. [Google Scholar] [PubMed]
- Guo, J.; Otis, J.M.; Higginbotham, H.; Monckton, C.; Cheng, J.; Asokan, A.; Mykytyn, K.; Caspary, T.; Stuber, G.D.; Anton, E. Primary Cilia Signaling Shapes the Development of Interneuronal Connectivity. Dev. Cell 2017, 42, 286–300.e4. [Google Scholar] [CrossRef] [PubMed]
- Bowie, E.; Goetz, S.C. TTBK2 and primary cilia are essential for the connectivity and survival of cerebellar Purkinje neurons. eLife 2020, 9, e51166. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, B.; Chen, C.; Li, C.; Zhang, Y. 5-HT6R null mutatrion induces synaptic and cognitive defects. Aging Cell 2021, 20, e13369. [Google Scholar] [CrossRef]
- Han, Y.-G.; Alvarez-Buylla, A. Role of primary cilia in brain development and cancer. Curr. Opin. Neurobiol. 2010, 20, 58–67. [Google Scholar] [CrossRef]
- Youn, Y.H.; Han, Y.-G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef]
- Feldman, D.; Banerjee, A.; Sur, M. Developmental Dynamics of Rett Syndrome. Neural Plast. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Stefan, H.; Lopes da Silva, F.H. Epileptic neuronal networks: Methods of identification and clinical relevance. Front. Neurol. 2013, 4, 8. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Tereshko, L.; Gao, Y.; Cary, B.A.; Turrigiano, G.G.; Sengupta, P. Ciliary neuropeptidergic signaling dynamically regulates excitatory synapses in postnatal neocortical pyramidal neurons. eLife 2021, 10, e65427. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-H.; Upadhyayula, S.; Dupuy, V.; Pang, S.; Deng, F.; Wan, J.; Walpita, D.; Pasolli, H.A.; Houser, J.; Sanchez-Martinez, S.; et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 2022, 185, 3390–3407.e18. [Google Scholar] [CrossRef] [PubMed]
- Wachten, D.; Mick, D.U. Signal transduction in primary cilia–analyzing and manipulating GPCR and second messenger signaling. Pharmacol. Ther. 2021, 224, 107836. [Google Scholar] [CrossRef]
- Barbeito, P.; Garcia-Gonzalo, F.R. HTR6 and SSTR3 targeting to primary cilia. Biochem. Soc. Trans. 2021, 49, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Händel, M.; Schulz, S.; Stanarius, A.; Schreff, M.; Erdtmann-Vourliotis, M.; Schmidt, H.; Wolf, G.; Höllt, V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 1999, 89, 909–926. [Google Scholar] [CrossRef]
- Diniz, G.B.; Battagello, D.S.; Klein, M.O.; Bono, B.S.M.; Ferreira, J.G.P.; Motta-Teixeira, L.C.; Gygi, D. Ciliary melanin-concentrating hormone receptor 1 (MCHR1) is widely distributed in the murine CNS in a sex-independent manner. J. Neurosci. Res. 2020, 98, 2045–2071. [Google Scholar] [CrossRef]
- Qiu, L.; LeBel, R.P.; Storm, D.R.; Chen, X. Type 3 adenylyl cyclase: A key enzyme mediating the cAMP signaling in neuronal cilia. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 95–108. [Google Scholar]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Kohli, P.; Höhne, M.; Jüngst, C.; Bertsch, S.; Ebert, L.K.; Schauss, A.C.; Benzing, T.; Rinschen, M.M.; Schermer, B. The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep. 2017, 18, 1521–1535. [Google Scholar] [CrossRef]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef]
- May, E.A.; Kalocsay, M.; D’Auriac, I.G.; Schuster, P.S.; Gygi, S.P.; Nachury, M.V.; Mick, D.U. Time-resolved proteomics profiling of the ciliary Hedgehog response. J. Cell Biol. 2021, 220, e202007207. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, Z.; Yang, F.; Zhou, T.; Xie, S. Deciphering cilia and ciliopathies using proteomic approaches. FEBS J. 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yam, P.T.; Chen, W.; Schlienger, S.; Gutierrez, O.T.; Cai, E.; Klein, G. Cilium proteomics reveals Numb as a positive regulator of the Hedgehog signaling pathway. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hansen, J.N.; Kaiser, F.; Klausen, C.; Stüven, B.; Chong, R.; Bönigk, W.; Mick, D.U.; Möglich, A.; Jurisch-Yaksi, N.; Schmidt, F.I.; et al. Nanobody-directed targeting of optogenetic tools to study signaling in the primary cilium. eLife 2020, 9, e57907. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Chiba, S.; Nakayama, K. Practical method for superresolution imaging of primary cilia and centrioles by expansion microscopy using an amplibody for fluorescence signal amplification. Mol. Biol. Cell 2020, 31, 2195–2206. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
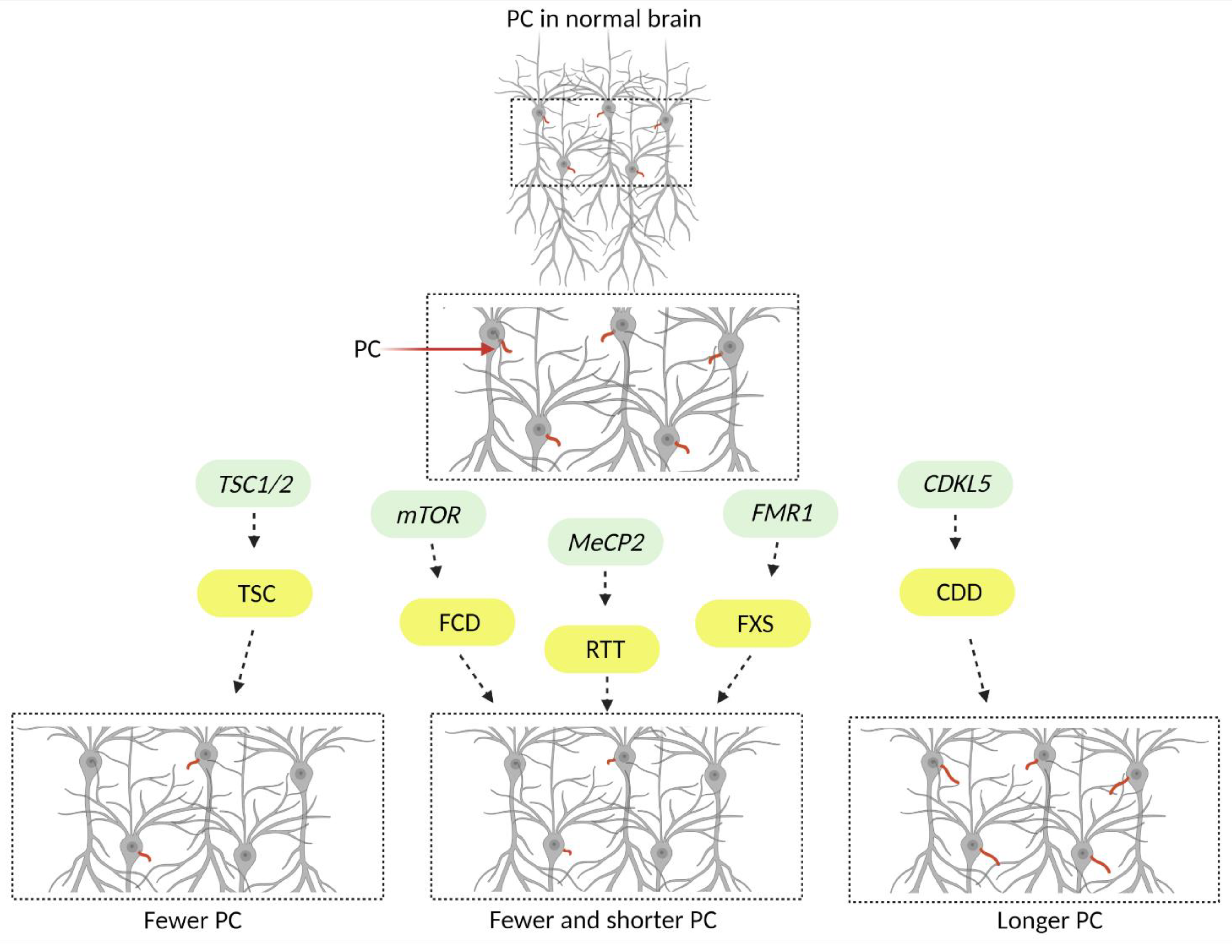
| Mutated Gene | Disease | Key Neurologic Features * | Experimental Systems + | Neuronal PC Phenotypes + | Ref |
|---|---|---|---|---|---|
| FMR1 | Fragile X Syndrome (FXS) | ASD, ID, seizures, ADHD, neuroanatomical abnormalities, i.e., larger volume of lateral ventricles |
| ↓ Number ↓ Length | [36,37,38] |
| TSC1/2 | Tuberous Sclerosis Complex (TSC) | Tubers, SENs, SEGAs, epilepsy, disorganized WM, ID, ASD, ADHD |
| ↓ Number | [39,40,41] |
| mTOR | Focal Cortical Dysplasia (FCD) | Epilepsy, ID, ASD, altered cortical architecture |
| ↓ Number ↓ Length | [42,43,44] |
| CDKL5 | CDKL5 Deficiency Disorder (CDD) | Infantile spasms, ASD, epilepsy, ID |
| ↑ Length | [45,46,47] |
| MeCP2 | Rett Syndrome (RTT) | ID, ASD, seizures |
| ↓ Number ↓ Length | [48,49] |
| Mouse Model | Primary Cilia Phenotypes * | Neurological Phenotypes + | Behavioral Phenotypes + | Ref |
|---|---|---|---|---|
| Mchr1Neo/Neo | n/a |
| Impaired learning and memory | [99] |
| Dnchc2 mutant | Structurally impaired in neuroectoderm |
| n/a | [100] |
| hGFAP-Cre; Kif3afl/fl | Loss from granule neuron precursors in DG |
| n/a | [26] |
| Emx1-Cre; Shhfl/- Emx1-Cre; Smofl/- | n/a |
| n/a | [101] |
| Cobblestone (hypomorphic Ift88) | Normal morphology in ventricles |
| n/a | [102] |
| Sst3 knockout | Normal in CA1 |
| Impaired recognition memory | [103] |
| Ift20fl/fl::mGFAP-Cre | Loss from radial neural stem cells in the SGZ |
| Delay in learning and enhanced cue-based fear responses | [104] |
| Ftm mutant |
Loss from telencephalic neuroepithelial cells |
| n/a | [105] |
| Ac3 mutant | Structurally intact in hippocampus | n/a | Impaired learning and memory | [106] |
| Arl13bfl/fl; Nex-Cre | n/a |
| n/a | [107] |
| Arl13bfl/fl; Dlx5/6-CIE | Shorter on interneurons |
| n/a | [107] |
| Inducible dominant negative Kif3a expressed in the hilus region of DG of adult mice | Loss from newborn DGCs |
| n/a | [108] |
| Arl13bhnn/hnn (null allele) | Shorter with disrupted morphological plasticity on radial progenitors in cortex |
| n/a | [109] |
| Ift20fl/fl AAV-CaMKII-eGFP-Cre injected in DG of adult mice | Loss from majority of GFP+ cells in DG |
| Impaired memory | [110] |
| Cobblestone (hypomorphic Ift88) | Normal morphology in VZ of ventral midbrain |
| n/a | [111] |
| Nestin-Kif3afl/fl | Loss from cortical ventricular surface and somatosensory cortex |
| n/a | [112] |
| Nkx2.1Cre; Arl13bfl/fl | Defective intraciliary Ca2+ signaling in striatal interneurons |
| n/a | [113] |
| Ttbk2fl/fl; Ubc-Cre-ERT2+ (Tamoxifen on P21) | Loss from cerebellum, brainstem, hippocampus, and cortex |
| Locomotor deficiencies | [114] |
| 5-ht6r mutant | Normal morphology in hippocampus |
| Anxiety and cognitive impairments | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karalis, V.; Donovan, K.E.; Sahin, M. Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies. J. Dev. Biol. 2022, 10, 54. https://doi.org/10.3390/jdb10040054
Karalis V, Donovan KE, Sahin M. Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies. Journal of Developmental Biology. 2022; 10(4):54. https://doi.org/10.3390/jdb10040054
Chicago/Turabian StyleKaralis, Vasiliki, Kathleen E. Donovan, and Mustafa Sahin. 2022. "Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies" Journal of Developmental Biology 10, no. 4: 54. https://doi.org/10.3390/jdb10040054
APA StyleKaralis, V., Donovan, K. E., & Sahin, M. (2022). Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies. Journal of Developmental Biology, 10(4), 54. https://doi.org/10.3390/jdb10040054