
Cleft Palate in Apert Syndrome



Abstract
1. Introduction
2. Materials and Methods
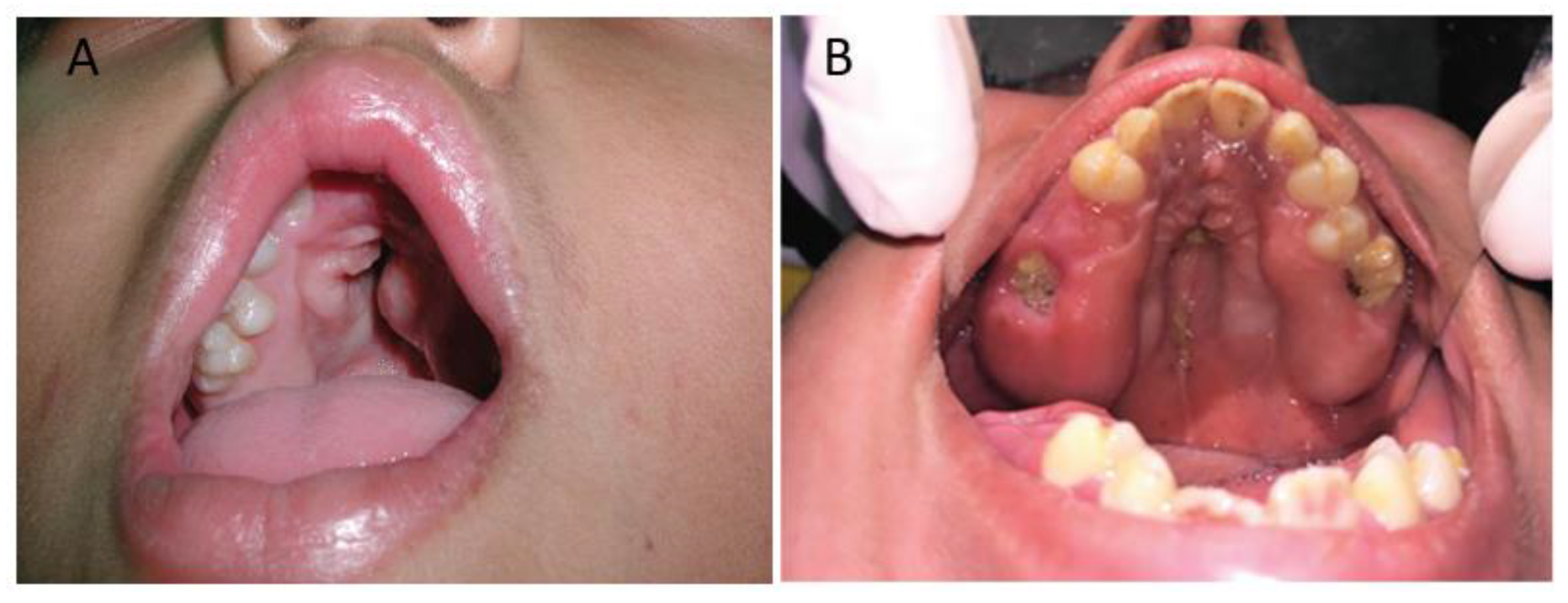
3. Clinical Characteristics of the Palate in Apert Syndrome
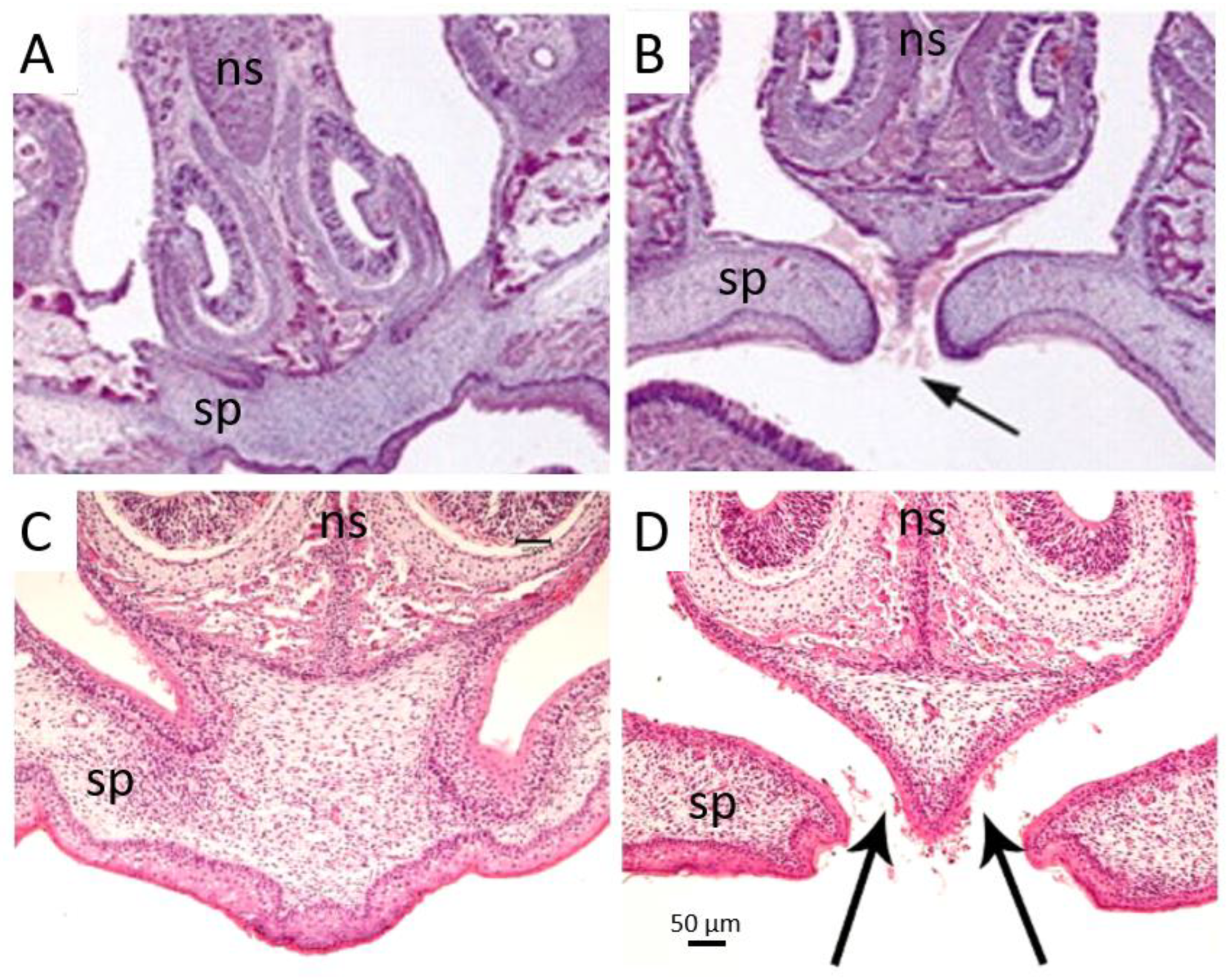
4. Palatal Phenotypes in Mouse Models of Apert Syndrome
5. Molecular and Cellular Mechanisms
5.1. FGFR2 Signaling
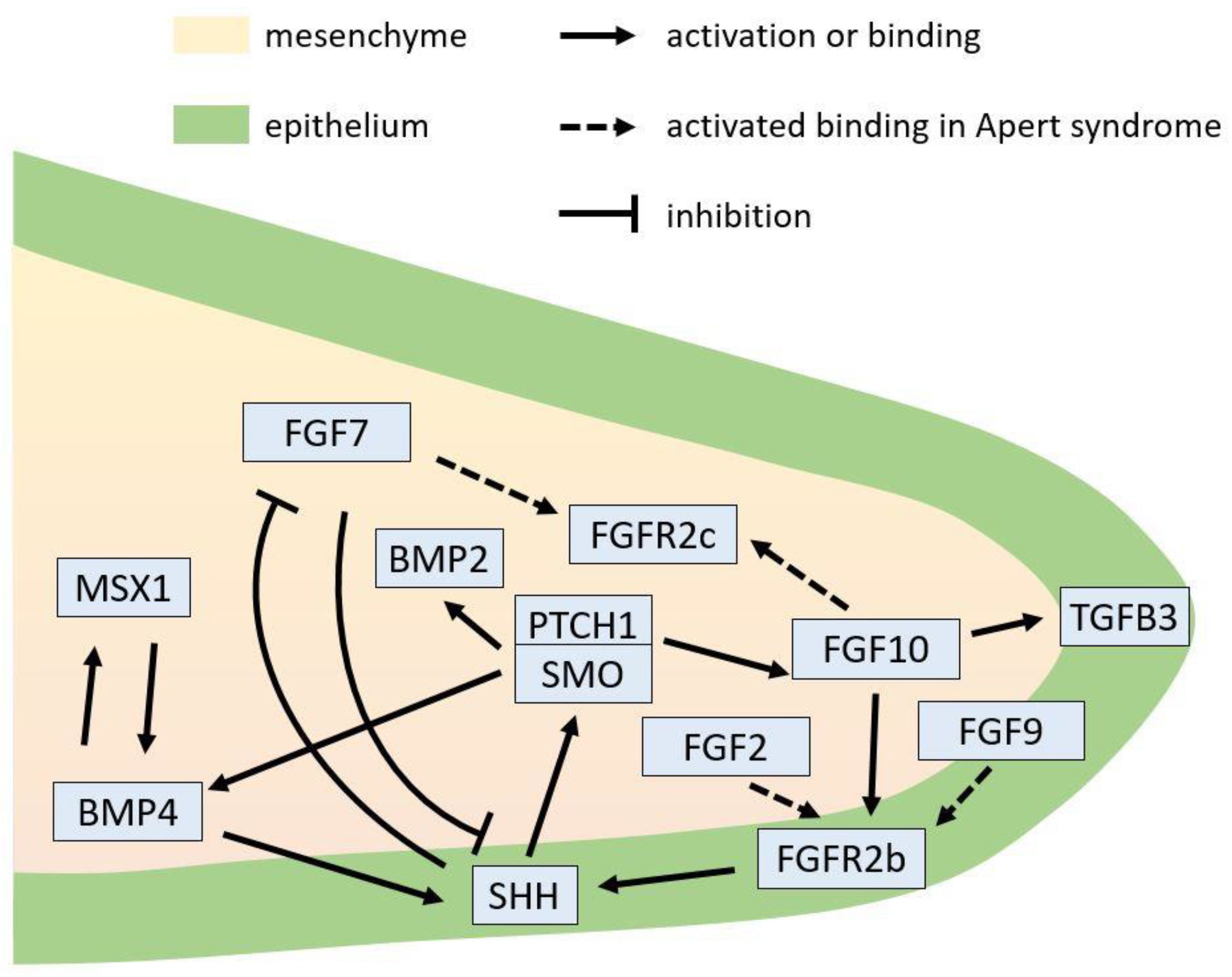
5.2. FGFR2-Related Signaling Network in Epithelial–Mesenchymal Interactions during Palatogenesis
6. Open Questions and Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nasreddine, G.; El Hajj, J.; Ghassibe-Sabbagh, M. Orofacial clefts embryology, classification, epidemiology, and genetics. Mutat. Res.-Rev. Mutat. Res. 2021, 787, 108373. [Google Scholar] [CrossRef] [PubMed]
- Wehby, G.L.; Cassell, C.H. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral Dis. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Mortensen, P.B. Facial clefting and psychiatric diseases: A follow-up of the Danish 1936–1987 facial cleft cohort. Cleft Palate-Craniofac. J. 2002, 39, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.T.; Cassell, C.H.; Meyer, R.E.; Isenburg, J.; Canfield, M.A.; Rickard, R.; Olney, R.S.; Stallings, E.B.; Beck, M.; Hashmi, S.S.; et al. Birth defects data from population-based birth defects surveillance programs in the United States, 2007 to 2011: Highlighting orofacial clefts. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, M.; Palmieri, A.; Carinci, F.; Scapoli, L. Non-syndromic cleft palate: An overview on human genetic and environmental risk factors. Front. Cell Dev. Biol. 2020, 8, 592271. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Bian, Z.; Torensma, R.; Von Den Hoff, J.W. Biological mechanisms in palatogenesis and cleft palate. J. Dent. Res. 2009, 88, 22–33. [Google Scholar] [CrossRef]
- Burg, M.L.; Chai, Y.; Yao, C.A.; Magee, W.; Figueiredo, J.C. Epidemiology, etiology, and treatment of isolated cleft palate. Front. Physiol. 2016, 7, 67. [Google Scholar] [CrossRef]
- Wenger, T.; Hing, A.; Evans, K. Apert syndrome. In GeneReviews® [Internet]; Adam, M., Mirzaa, G., Pagon, R., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2022; 2009. [Google Scholar]
- Blank, C.E. Apert’s syndrome (a type of acrocephalosyndactyly)–observations on a British series of thirty-nine cases. Ann. Hum. Genet. 1959, 24, 151–164. [Google Scholar] [CrossRef]
- Cohen, M.M.; Kreiborg, S.; Lammer, E.J.; Cordero, J.F.; Mastroiacovo, P.; Erickson, J.D.; Roeper, P.; Martinez-Frias, M.L. Birth prevalence study of the Apert syndrome. Am. J. Med. Genet. 1992, 42, 655–659. [Google Scholar] [CrossRef]
- Tolarova, M.M.; Harris, J.A.; Ordway, D.E.; Vargervik, K. Birth prevalence, mutation rate, sex ratio, parents’ age, and ethnicity in Apert syndrome. J. Med. Genet. 1997, 72, 394–398. [Google Scholar] [CrossRef]
- Dionne, C.A.; Crumley, G.; Bellot, F.; Kaplow, J.M.; Searfoss, G.; Ruta, M.; Burgess, W.H.; Jaye, M.; Schlessinger, J. Cloning and expression of two distinct high-affinity receptors cross-reacting with acidic and basic fibroblast growth factors. EMBO J. 1990, 9, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1992, 60, 1–41. [Google Scholar]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Wilkie, A.O.M.; Slaney, S.F.; Oldridge, M.; Poole, M.D.; Ashworth, G.J.; Hockley, A.D.; Hayward, R.D.; David, D.J.; Pulleyn, L.J.; Rutland, P.; et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 1995, 9, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Park, W.J.; Theda, C.; Maestri, N.E.; Meyers, G.A.; Fryburg, J.S.; Dufresne, C.; Cohen, M.M.; Jabs, E.W. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am. J. Hum. Genet. 1995, 57, 321–328. [Google Scholar] [PubMed]
- Lajeunie, E.; Cameron, R.; El Ghouzzi, V.; De Parseval, N.; Journeau, P.; Gonzales, M.; Delezoide, A.L.; Bonaventure, J.; Le Merrer, M.; Renier, D. Clinical variability in patients with Apert’s syndrome. J. Neurosurg. 1999, 90, 443–447. [Google Scholar] [CrossRef]
- Goriely, A.; McVean, G.A.T.; Van Pelt, A.M.M.; O’Rourke, A.W.; Wall, S.A.; De Rooij, D.G.; Wilkie, A.O.M. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc. Natl. Acad. Sci. USA 2005, 102, 6051–6056. [Google Scholar] [CrossRef]
- Goriely, A.; McVean, G.A.T.; Röjmyr, M.; Ingemarsson, B.; Wilkie, A.O.M. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 2003, 301, 643–646. [Google Scholar] [CrossRef]
- Oldridge, M.; Lunt, P.W.; Zackai, E.H.; McDonald-McGinn, D.M.; Muenke, M.; Moloney, D.M.; Twigg, S.R.F.; Heath, J.K.; Howard, T.D.; Hoganson, G.; et al. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum. Mol. Genet. 1997, 6, 137–143. [Google Scholar] [CrossRef]
- Shi, Q.; Dai, R.; Wang, R.; Jing, J.; Yu, X.; Liu, R.; Liu, Y. A novel FGFR2 (S137W) mutation resulting in Apert syndrome: A case report. Medicine 2020, 99, e22340. [Google Scholar] [CrossRef]
- Oldridge, M.; Zackai, E.H.; McDonald-McGinn, D.M.; Iseki, S.; Morriss-Kay, G.M.; Twigg, S.R.F.; Johnson, D.; Wall, S.A.; Jiang, W.; Theda, C.; et al. De novo Alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am. J. Hum. Genet. 1999, 64, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, A.L.; Bowdin, S.C.; Klatt, R.E.M.; Wilkie, A.O.M. A deletion of FGFR2 creating a chimeric IIIb/IIIc exon in a child with Apert syndrome. BMC Med. Genet. 2011, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, O.A.; Eliseenkova, A.V.; Plotnikov, A.N.; Yu, K.; Ornitz, D.M.; Mohammadi, M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. USA 2001, 98, 7182–7187. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Herr, A.B.; Waksman, G.; Ornitz, D.M. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 14536–14541. [Google Scholar] [CrossRef]
- Lan, Y.; Xu, J.; Jiang, R. Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 2015, 115, 59–84. [Google Scholar]
- Kosowski, T.; Weathers, W.; Wolfswinkel, E.; Ridgway, E. Cleft palate. Semin. Plast. Surg. 2012, 26, 164–169. [Google Scholar] [CrossRef]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 231–243. [Google Scholar] [CrossRef]
- Gritli-Linde, A. Molecular control of secondary palate development. Dev. Biol. 2007, 301, 309–326. [Google Scholar] [CrossRef]
- Lane, J.; Kaartinen, V. Signaling networks in palate development. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 271–278. [Google Scholar] [CrossRef]
- Alam, M.K.; Alfawzan, A.A.; Srivastava, K.C.; Shrivastava, D.; Ganji, K.K.; Manay, S.M. Craniofacial morphology in Apert syndrome: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 5708. [Google Scholar] [CrossRef]
- Kreiborg, S.; Cohen, M.M., Jr. The oral manifestations of Apert syndrome. J. Craniofac. Genet. Dev. Biol. 1992, 12, 41–48. [Google Scholar] [PubMed]
- Kobayashi, Y.; Ogura, K.; Hikita, R.; Tsuji, M.; Moriyama, K. Craniofacial, oral, and cervical morphological characteristics in Japanese patients with Apert syndrome or Crouzon syndrome. Eur. J. Orthod. 2021, 43, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Hohoff, A.; Joos, U.; Meyer, U.; Ehmer, U.; Stamm, T. The spectrum of Apert syndrome: Phenotype, particularities in orthodontic treatment, and characteristics of orthognathic surgery. Head Face Med. 2007, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Letra, A.; de Almeida, A.L.P.F.; Kaizer, R.; Esper, L.A.; Sgarbosa, S.; Granjeiro, J.M. Intraoral features of Apert’s syndrome. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2007, 103, e38–e41. [Google Scholar] [CrossRef]
- Solomon, L.M.; Medenica, M.; Pruzansky, S.; Kreiborg, S. Apert syndrome and palatal mucopolysaccharides. Teratology 1973, 8, 287–291. [Google Scholar] [CrossRef]
- Peterson, S.J.; Pruzansky, S. Palatal anomalies in the syndromes of Apert and Crouzon. Cleft Palate J. 1974, 11, 394–403. [Google Scholar] [CrossRef]
- Peterson-Falzone, S.J.; Pruzansky, S.; Parris, P.J.; Laffer, J.L. Nasopharyngeal dysmorphology in the syndromes of Apert and Crouzon. Cleft Palate J. 1981, 18, 237–250. [Google Scholar]
- Cohen, M.M.; Kreiborg, S. A clinical study of the craniofacial features in Apert syndrome. Int. J. Oral Maxillofac. Surg. 1996, 25, 45–53. [Google Scholar] [CrossRef]
- Arroyo Carrera, I.; Martínez-Frías, M.L.; Marco Pérez, J.J.; Paisán Grisolía, L.; Cárdenes Rodríguez, A.; Nieto Conde, C.; Félix Rodríguez, V.; Egüés Jimeno, J.J.; Morales Fernández, M.C.; Gómez-Ullate Vergara, J.; et al. Apert syndrome: Clinico-epidemiological analysis of a series of consecutive cases in Spain. An. Esp. Pediatr. 1999, 51, 667–672. [Google Scholar]
- Albuquerque, M.A.P.; Cavalcanti, M.G.P. Computed tomography assessment of Apert syndrome. Braz. Oral Res. 2004, 18, 35–39. [Google Scholar] [CrossRef]
- Stavropoulos, D.; Tarnow, P.; Mohlin, B.; Kahnberg, K.E.; Hagberg, C. Comparing patients with Apert and Crouzon syndromes-clinical features and cranio-maxillofacial surgical reconstruction. Swed. Dent. J. 2012, 36, 25–34. [Google Scholar] [PubMed]
- Kakutani, H.; Sato, Y.; Tsukamoto-Takakusagi, Y.; Saito, F.; Oyama, A.; Iida, J. Evaluation of the maxillofacial morphological characteristics of Apert syndrome infants. Congenit. Anom. 2017, 57, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Kobayashi, Y.; Hikita, R.; Tsuji, M.; Moriyama, K. Three-dimensional analysis of the palatal morphology in growing patients with Apert syndrome and Crouzon syndrome. Congenit. Anom. 2022, 62, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Duggal, R.; Parkash, H. Dentofacial characteristics in Apert syndrome: A case report. J. Indian Soc. Pedod. Prev. Dent. 2002, 20, 118–123. [Google Scholar] [PubMed]
- Vijayalakshmi, A.M.; Menon, A. Apert syndrome. Indian Pediatr. 2002, 39, 876–878. [Google Scholar] [PubMed]
- Huang, F.; Sweet, R.; Tewfik, T.L. Apert syndrome and hearing loss with ear anomalies: A case report and literature review. Int. J. Pediatr. Otorhinolaryngol. 2004, 68, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Draznin, M. Apert syndrome. Dermatol. Online J. 2005, 11, 53–56. [Google Scholar] [CrossRef]
- Tosun, G.; Sener, Y. Apert syndrome with glucose-6-phosphate dehydrogenase deficiency: A case report. Int. J. Paediatr. Dent. 2006, 16, 218–221. [Google Scholar] [CrossRef]
- Herman, T.E.; Siegel, M.J. Apert syndrome with omphalocele. J. Perinatol. 2010, 30, 695–697. [Google Scholar] [CrossRef]
- Premalatha; Kannan, V.P.; Madhu. Apert syndrome. J. Indian Soc. Pedod. Prev. Dent. 2010, 28, 322–325. [Google Scholar]
- Şoancă, A.; Dudea, D.; Gocan, H.; Roman, A.; Culic, B. Oral manifestations in Apert syndrome: Case presentation and a brief review of the literature. Rom. J. Morphol. Embryol. 2010, 51, 581–584. [Google Scholar] [PubMed]
- Vadiati Saberi, B.; Shakoorpour, A. Apert syndrome: Report of a case with emphasis on oral manifestations. J. Dent. 2011, 8, 90–95. [Google Scholar]
- Costa, F.W.G.; Rodrigues, R.R.; Batista, A.C.B.; Ribeiro, T.R.; Pereira, K.M.A. Multiple radiopaque mandibular lesions in a patient with Apert syndrome. J. Endod. 2012, 38, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Ileri, Z.; Goyenc, Y.B. Apert syndrome: A case report. Eur. J. Dent. 2012, 6, 110. [Google Scholar] [CrossRef][Green Version]
- Khan, S.; Chatra, L.; Shenai, P.; Veena, K. Apert syndrome: A case report. Int. J. Clin. Pediatr. Dent. 2012, 5, 203. [Google Scholar]
- Bhatia, P.; Patel, P.; Jani, Y.; Soni, N. Apert’s syndrome: Report of a rare case. J. Oral Maxillofac. Pathol. 2013, 17, 294–297. [Google Scholar]
- Aggarwal, H.; Singh, S.V.; Kumar, P. Apert syndrome: A rare anomalad. CHRISMED J. Health Res. 2014, 1, 206. [Google Scholar] [CrossRef]
- Ercoli, G.; Bidondo, M.P.; Senra, B.C.; Groisman, B. Apert syndrome with omphalocele: A case report. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 726–729. [Google Scholar] [CrossRef]
- Kumar, G.R.; Jyothsna, M.; Ahmed, S.B.; Lakshmi, K.R.S. Apert’s syndrome. Int. J. Clin. Pediatr. Dent. 2014, 7, 72. [Google Scholar]
- Spruijt, B.; Rijken, B.F.M.; Joosten, K.F.M.; Bredero-Boelhouwer, H.H.; Pullens, B.; Lequin, M.H.; Wolvius, E.B.; van Veelen-Vincent, M.L.C.; Mathijssen, I.M.J. Atypical presentation of a newborn with Apert syndrome. Child’s Nerv. Syst. 2015, 31, 481–486. [Google Scholar] [CrossRef]
- Barman, A.; Dutta, B.C.; Sarkar, J.K. Apert syndrome: A case report. Natl. J. Clin. Anat. 2015, 4, 145–148. [Google Scholar] [CrossRef]
- Torres, L.; Hernández, G.; Barrera, A.; Ospina, S.; Prada, R. Molecular analysis of exons 8, 9 and 10 of the fibroblast growth factor receptor 2 (FGFR2) gene in two families with index cases of Apert syndrome. Colomb. Med. 2015, 46, 150–153. [Google Scholar] [CrossRef]
- Işık, E.; Atik, T.; Onay, H.; Özkınay, F. Two patients with Apert syndrome with different mutations: The importance of early diagnosis. Turk Pediatr. Ars. 2017, 52, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.K.; Choi, D.S.; Jang, I.S.; Yook, H.T.; Lee, S.Y.; Lee, S.S.; Lee, S.K. Aberrant growth of the anterior cranial base relevant to severe midface hypoplasia of Apert syndrome. Maxillofac. Plast. Reconstr. Surg. 2018, 40, 40. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Ouedraogo, Y.S.; Nacro, F.S.; Sanogo, B.; Kombasséré, S.O.; Ouermi, A.S.; Tamboura, H.; Cessouma, R.K.; Nacro, B. Apert syndrome: Diagnostic and management problems in a resource-limited country. Pediatr. Rep. 2019, 11, 78–80. [Google Scholar] [CrossRef]
- Brajadenta, G.S.; Sari, A.I.P.; Nauphar, D.; Pratamawati, T.M.; Thoreau, V. Molecular analysis of exon 7 of the fibroblast growth factor receptor 2 (FGFR2) gene in an Indonesian patient with Apert syndrome: A case report. J. Med. Case Rep. 2019, 13, 244. [Google Scholar] [CrossRef]
- Cammarata-Scalisi, F.; Yilmaz, E.; Callea, M.; Avendaño, A.; Mıhçı, E.; Alper, O.M. Clinical and genetic findings of two cases with Apert syndrome. Bol. Med. Hosp. Infant. Mex. 2019, 76, 44–48. [Google Scholar] [CrossRef]
- Dap, M.; Bach-Segura, P.; Bertholdt, C.; Menzies, D.; Masutti, J.P.; Klein, O.; Perdriolle-Galet, E.; Lambert, L.; Morel, O. Variable phenotypic expression of Apert syndrome in monozygotic twins. Clin. Case Rep. 2019, 7, 54–57. [Google Scholar] [CrossRef]
- Kumar, G.; Garg, A.; Vignesh, R.; Dhillon, J.K.; Faraz, F. Apert syndrome: A case report. J. South Asian Assoc. Pediatr. Dent. 2019, 2, 32–34. [Google Scholar]
- Chavda, V.; Shah, A.; Chaudhari, D. Acrocephalosyndactyly type 1 (Apert syndrome): A case report. Indian Dermatol. Online J. 2021, 12, 958. [Google Scholar] [CrossRef]
- Jose, B.; Emmatty, T.B.; Methippara, J.J.; Kumar, K.; Thampi, N.M. Apert syndrome: An insight into dentofacial features. Cureus 2021, 13, e17735. [Google Scholar] [CrossRef] [PubMed]
- Tonni, G.; Grisolia, G.; Baldi, M.; Bonasoni, M.P.; Ginocchi, V.; Rolo, L.C.; Araujo Júnior, E. Early prenatal ultrasound and molecular diagnosis of Apert syndrome: Case report with postmortem CT-scan and chondral plate histology. Fetal Pediatr. Pathol. 2022, 41, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Samanta, S. Bifid uvula: Anesthetist don’t take it lightly! Saudi J. Anaesth. 2013, 7, 482. [Google Scholar] [CrossRef]
- Reiter, R.; Brosch, S.; Wefel, H.; Schlömer, G.; Haase, S. The submucous cleft palate: Diagnosis and therapy. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.K.; Conley, F.S.; Marks, S.; Larson, D.L. Submucous cleft palate: Diagnostic methods and outcomes of surgical treatment. Plast. Reconstr. Surg. 1996, 97, 1497–1509. [Google Scholar] [CrossRef]
- Cohen, M.M.; Kreiborg, S. Upper and lower airway compromise in the Apert syndrome. Am. J. Med. Genet. 1992, 44, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Slaney, S.F.; Oldridge, M.; Hurst, J.A.; Morriss-Kay, G.M.; Hall, C.M.; Poole, M.D.; Wilkie, A.O.M. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet. 1996, 58, 923–932. [Google Scholar]
- Von Gernet, S.; Golla, A.; Ehrenfels, Y.; Schuffenhauer, S.; Fairley, J.D. Genotype–phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin. Genet. 2000, 57, 137–139. [Google Scholar] [CrossRef]
- Kilcoyne, S.; Luscombe, C.; Scully, P.; Overton, S.; Brockbank, S.; Swan, M.C.; Johnson, D.; Wall, S.; Wilkie, A.O.M. Hearing, speech, language, and communicative participation in patients with Apert syndrome: Analysis of correlation with fibroblast growth factor receptor 2 mutation. J. Craniofac. Surg. 2022, 33, 243–250. [Google Scholar] [CrossRef]
- Sakai, N.; Tokunaga, K.; Yamazaki, Y.; Shida, H.; Sakata, Y.; Susami, T.; Nakakita, N.; Takato, T.; Uchinuma, E. Sequence analysis of fibroblast growth factor receptor 2 (FGFR2) in Japanese patients with craniosynostosis. J. Craniofac. Surg. 2001, 12, 580–585. [Google Scholar] [CrossRef]
- Holmes, G. Mouse models of Apert syndrome. Child’s Nerv. Syst. 2012, 28, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, D.; Li, C.; Engel, A.; Deng, C.X. A Ser250Trp substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 2003, 33, 169–178. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, R.; Yang, F.; Karim, B.O.; Iacovelli, A.J.; Cai, J.; Lerner, C.P.; Ricthsmeier, J.T.; Leszl, J.M.; Hill, C.A.; et al. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2+/S252W mouse. Development 2005, 132, 3537–3548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, M.; Uhlhorn, V.L.; Zhou, X.; Peter, I.; Martinez-Abadias, N.; Hill, C.A.; Percival, C.J.; Richtsmeier, J.T.; Huso, D.L.; et al. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2 + P253R mice. BMC Dev. Biol. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Du, X.; Li, C.; Xu, X.; Chen, Z.; Su, N.; Zhao, L.; Qi, H.; Li, F.; Xue, J.; et al. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone 2008, 42, 631–643. [Google Scholar] [CrossRef]
- Hajihosseini, M.K.; Wilson, S.; De Moerlooze, L.; Dickson, C. A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffersyndrome-like phenotypes. Proc. Natl. Acad. Sci. USA 2001, 98, 3855–3860. [Google Scholar] [CrossRef]
- Miki, T.; Bottaro, D.P.; Fleming, T.P.; Smith, C.L.; Burgess, W.H.; Chan, A.M.L.; Aaronson, S.A. Determination of ligand-binding specificity by alternative splicing: Two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. USA 1992, 89, 246–250. [Google Scholar] [CrossRef]
- Orr-Urtreger, A.; Bedford, M.T.; Burakova, T.; Arman, E.; Zimmer, Y.; Yayon, A.; Givol, D.; Lonai, P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev. Biol. 1993, 158, 475–486. [Google Scholar] [CrossRef]
- Holmes, G.; Basilico, C. Mesodermal expression of Fgfr2S252W is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev. Biol. 2012, 368, 283–293. [Google Scholar] [CrossRef]
- Martínez-Abadías, N.; Holmes, G.; Pankratz, T.; Wang, Y.; Zhou, X.; Jabs, E.W.; Richtsmeier, J.T. From shape to cells: Mouse models reveal mechanisms altering palate development in Apert syndrome. Dis. Model. Mech. 2013, 6, 768–779. [Google Scholar] [CrossRef]
- Yu, K.; Karuppaiah, K.; Ornitz, D.M. Mesenchymal fibroblast growth factor receptor signaling regulates palatal shelf elevation during secondary palate formation. Dev. Dyn. 2015, 244, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Chen, Z.; Xiao, Q.; Li, R.; Chen, Z. A review of FGF signaling in palate development. Biomed. Pharmacother. 2018, 103, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Porntaveetus, T.; Oommen, S.; Sharpe, P.T.; Ohazama, A. Expression of Fgf signalling pathway related genes during palatal rugae development in the mouse. Gene Expr. Patterns 2010, 10, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Burns, H.D.; Enriquez-Harris, P.; Wilkie, A.O.M.; Heath, J.K. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum. Mol. Genet. 1998, 7, 1475–1483. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar] [CrossRef]
- Igarashi, M.; Finch, P.W.; Aaronson, S.A. Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7). J. Biol. Chem. 1998, 273, 13230–13235. [Google Scholar] [CrossRef]
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of Fgf signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef]
- Ray, A.T.; Mazot, P.; Richard Brewer, J.; Catela, C.; Dinsmore, C.J.; Soriano, P. FGF signaling regulates development by processes beyond canonical pathways. Genes Dev. 2020, 34, 1735–1752. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.; Han, D.; Grimaldi, A.; Sarrion, P.; Park, S.S.; Pelikan, R.; Sanchez-Lara, P.A.; Chai, Y. Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Development 2015, 142, 3734–3745. [Google Scholar] [CrossRef]
- Perrine, S.M.M.; Wu, M.; Holmes, G.; Bjork, B.C.; Jabs, E.W.; Richtsmeier, J.T. Phenotypes, developmental basis, and genetics of Pierre Robin complex. J. Dev. Biol. 2020, 8, 30. [Google Scholar] [CrossRef]
- Matsumura, K.; Taketomi, T.; Yoshizaki, K.; Arai, S.; Sanui, T.; Yoshiga, D.; Yoshimura, A.; Nakamura, S. Sprouty2 controls proliferation of palate mesenchymal cells via fibroblast growth factor signaling. Biochem. Biophys. Res. Commun. 2011, 404, 1076–1082. [Google Scholar] [CrossRef]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P.C. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef]
- Lan, Y.; Jiang, R. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 2009, 136, 1387–1396. [Google Scholar] [CrossRef]
- Economou, A.D.; Ohazama, A.; Porntaveetus, T.; Sharpe, P.T.; Kondo, S.; Basson, M.A.; Gritli-Linde, A.; Cobourne, M.T.; Green, J.B.A. Periodic stripe formation by a Turing mechanism operating at growth zones in the mammalian palate. Nat. Genet. 2012, 44, 348–351. [Google Scholar] [CrossRef]
- Hajihosseini, M.K.; Duarte, R.; Pegrum, J.; Donjacour, A.; Lana-Elola, E.; Rice, D.P.; Sharpe, J.; Dickson, C. Evidence that Fgf10 contributes to the skeletal and visceral defects of an Apert syndrome mouse model. Dev. Dyn. 2009, 238, 376–385. [Google Scholar] [CrossRef]
- Hosokawa, R.; Deng, X.; Takamori, K.; Xu, X.; Urata, M.; Bringas, B., Jr.; Chai, Y. Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312B, 343–350. [Google Scholar] [CrossRef]
- Han, J.; Mayo, J.; Xu, X.; Li, J.; Bringas, P.; Maas, R.L.; Rubenstein, J.L.R.; Chai, Y. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development 2009, 136, 4225–4233. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, Y.; Zhao, X.; Zhang, X.; Fermin, C.; Chen, Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002, 129, 4135–4146. [Google Scholar] [CrossRef]
- Alappat, S.R.; Zhang, Z.; Suzuki, K.; Zhang, X.; Liu, H.; Jiang, R.; Yamada, G.; Chen, Y. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 2005, 277, 102–113. [Google Scholar] [CrossRef]
- Britto, J.A.; Evans, R.D.; Hayward, R.D.; Jones, B.M. Toward pathogenesis of Apert cleft palate: FGF, FGFR, and TGFβ genes are differentially expressed in sequential stages of human palatal shelf fusion. Cleft Palate-Craniofac. J. 2002, 39, 332–340. [Google Scholar] [CrossRef]
- Xie, C.; De, S.; Selby, A. Management of the airway in Apert syndrome. J. Craniofac. Surg. 2016, 27, 137–141. [Google Scholar] [CrossRef]
- Shukla, V.; Coumoul, X.; Wang, R.H.; Kim, H.S.; Deng, C.X. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 2007, 39, 1145–1150. [Google Scholar] [CrossRef]
- Oliver, J.D.; Jia, S.; Halpern, L.R.; Graham, E.M.; Turner, E.C.; Colombo, J.S.; Grainger, D.W.; D’Souza, R.N. Innovative molecular and cellular therapeutics in cleft palate tissue engineering. Tissue Eng.-Part B Rev. 2021, 27, 215–237. [Google Scholar] [CrossRef]
- Mazzetti, M.P.V.; Alonso, N.; Brock, R.S.; Ayoub, A.; Massumoto, S.M.; Piñero Eça, L. Importance of stem cell transplantation in cleft lip and palate surgical treatment protocol. J. Craniofac. Surg. 2018, 29, 1445–1451. [Google Scholar] [CrossRef]
- Conley, Z.R.; Hague, M.; Kurosaka, H.; Dixon, J.; Dixon, M.J.; Trainor, P.A. A quantitative method for defining high-arched palate using the Tcof1+/− mutant mouse as a model. Dev. Biol. 2016, 415, 296–305. [Google Scholar] [CrossRef]
- Tabler, J.M.; Barrell, W.B.; Szabo-Rogers, H.L.; Healy, C.; Yeung, Y.; Perdiguero, E.G.; Schulz, C.; Yannakoudakis, B.Z.; Mesbahi, A.; Wlodarczyk, B.; et al. Fuz mutant mice reveal shared mechanisms between ciliopathies and FGF-related syndromes. Dev. Cell 2013, 25, 623–635. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Palatal Phenotypes | References |
|---|---|---|
| Solomon et al. (1973) | In a cohort of 13 patients, all 13 (100%) presented with a Byzantine palatal arch; 6 of 13 (46%) presented with a bifid uvula; 3 of 13 (23%) presented with a cleft of the soft palate. | [36] |
| Peterson; Pruzansky (1974) | In a cohort of 19 patients, all 19 (100%) presented with a narrow, high-arched palate with lateral accumulations of soft tissue barely separated by a deep median groove; 6 of 19 (32%) presented with a bifid uvula; 2 of 19 (11%) presented with a cleft palate. On radiographic examination, 10 of 19 (53%) presented with abnormal length of the soft palate, and 8 of 19 (42%) showed abnormal velar thickness (4 overlapping cases). | [37] |
| Peterson-Falzone et al. (1981) | In a cohort of 29 patients, alterations of the nasopharyngeal architecture were found. Hard palate length was reduced and soft palate length was greater than the norm. | [38] |
| Kreiborg; Cohen (1992) | In a cohort of 119 patients, almost all patients presented with a Byzantine arch-shaped palate; approximately 75% of patients presented with a cleft of the soft palate or bifid uvula. | [32] |
| Cohen; Kreiborg (1996) | In a cohort of 136 patients 1, almost all patients (94%) presented with a highly arched, constricted palate and median furrow. Lateral palatal swellings were present that increased in size with age. The hard palate was shorter than normal and the soft palate was both longer and thicker than normal. | [39] |
| Arroyo Carrera et al. (1999) | In a cohort of 17 patients, 4 of 17 (23.5%) presented with a cleft palate. | [40] |
| Albuquerque; Cavalcanti (2004) | In a cohort of 5 patients, all (100%) presented with a pseudo-cleft in the midline palate. | [41] |
| Letra et al. (2007) | In a cohort of 23 patients, 16 of 23 (70%) presented with an arched palate; 21 of 23 (91%) presented with lateral gingival swellings; 1 of 23 (4%) presented with a cleft of the soft palate. | [35] |
| Stavropoulos et al. (2012) | In a cohort of 23 patients with Apert syndrome and 28 patients with Crouzon syndrome, cleft palate and extensive lateral palatal soft tissue swellings were more common in children with Apert syndrome than Crouzon syndrome. | [42] |
| Kakutani et al. (2017) | In a cohort of 5 patients, all 5 (100%) had a pseudo-cleft palate with a Byzantine arch shape; 4 of 5 (80%) presented with narrowing in the upper arch. | [43] |
| Kobayashi et al. (2021) | In a cohort of 7 patients, all 7 (100%) had a high palate with lateral palatal swellings; 2 of 7 (28.6%) presented with a cleft of the soft palate; 1 of 7 (14.3%) presented with a cleft of the hard palate. | [33] |
| Ogura et al. (2022) | In a cohort of 4 patients, 2 of 4 (50%) presented with a cleft of the soft palate; 1 of 4 (25%) presented with a cleft of the hard palate. | [44] |
| Study | Palatal Phenotypes | References |
|---|---|---|
| Batra et al. (2002) | A female patient had a pseudo-cleft palate. | [45] |
| Vijayalakshmi; Menon (2002) | A male patient had a cleft of the soft palate. | [46] |
| Huang et al. (2004) | A female patient had a submucous cleft palate and absent uvula. | [47] |
| Verma et al. (2005) | A male patient had a cleft palate. | [48] |
| Tosun et al. (2006) | A male patient had a V-shaped maxillary arch with a midline pseudo-cleft and lateral swellings on the palatal process. | [49] |
| Herman; Siegel (2010) | A female patient with an S252W variant in the FGFR2 gene had a cleft of the soft palate. | [50] |
| Premalatha et al. (2010) | A male patient had a high-arched palate with a pseudo-cleft in the posterior one-third. | [51] |
| Şoancǎ et al. (2010) | A male patient had a Byzantine arch palate associated with lateral swellings of the palatine processes and a bifid uvula. | [52] |
| Vadiati Saberi; Shakoorpour (2011) | A female patient had an arched swelling (pseudo-cleft configuration) and a V-shaped maxillary arch. | [53] |
| Costa et al. (2012) | A female patient had a U-shaped dental arch, swelling of the lateral palatine processes on both sides, and a bifid uvula. | [54] |
| Ileri; Goyenc (2012) | A female patient had a cleft palate. | [55] |
| Khan et al. (2012) | A male patient had a V-shaped maxillary arch and a pseudo-cleft palate. | [56] |
| Bhatia et al. (2013) | A male patient had a deep pseudo-cleft. | [57] |
| Aggarwal et al. (2014) | A male patient had a bulky, high-arched V-shaped palate with an occult submucosal cleft and rotated maxillary and mandibular incisors. | [58] |
| Ercoli et al. (2014) | A male patient had a high-arched palate. | [59] |
| Kumar et al. (2014) | A male patient had a high-arched palate associated with lateral swellings of the palatine processes on either side of the midline, mimicking a pseudo-cleft. | [60] |
| Spruijt et al. (2015) | A male patient with an S252W variant in the FGFR2 gene had a high-arched, narrow palate. | [61] |
| Barman et al. (2015) | A male patient had a high-arched palate. | [62] |
| Torres et al. (2015) | A male patient had a high-arched palate. A variant NM_000141.5: c.939+42T>A (T78.501A) located near the splicing site in FGFR2 was found. | [63] |
| Işık et al. (2017) | A female patient had a cleft palate. | [64] |
| Cha et al. (2018) | A male patient had a narrow and triangular-shaped maxillary arch and Byzantine arch-shaped palate. | [65] |
| Barro et al. (2019) | A female patient had a cleft palate. | [66] |
| Brajadenta et al. (2019) | An Indonesian male patient with an S252W variant in the FGFR2 gene had maxillary hypoplasia with a high-arched palate. His V-shaped maxillary arch was filled with double rows of teeth. | [67] |
| Cammarata-Scalisi et al. (2019) | One of two unrelated female patients, one had a high-arched palate, and the other a cleft of the soft palate. In both patients, a heterozygous S252W variant was identified. | [68] |
| Dap et al. (2019) | One of two monozygotic twins with an S252W variant was found to have a cleft palate at 30 weeks of gestation. | [69] |
| Kumar et al. (2019) | A female patient had a high-arched palate, a pseudo-cleft, and gingival enlargement. | [70] |
| Chavda et al. (2021) | A male patient had a high-arched palate. | [71] |
| Jose et al. (2021) | A female patient had a pseudo-cleft. | [72] |
| Tonni et al. (2022) | A female fetus at 20 weeks of gestation was found to have a smooth palate with a midline cleft and an absent uvula. A heterozygous P253R variant was identified. | [73] |
| Study | Cohort | FGFR2 S252W | FGFR2 P253R | Notes | References | ||||
|---|---|---|---|---|---|---|---|---|---|
| Cohort Size | Abnormal Palate Number | % | Cleft Palate Fraction | Cleft Palate % | Cleft Palate Fraction | Cleft Palate % | |||
| Park et al. (1995) | 36 | Cleft palate in 16 patients | 44.4 | 15/16 | 93.8 | 1/16 | 6.2 | [16] | |
| Slaney et al. (1996) | 87 | Soft palate cleft or bifid uvula in 37 patients | 42.5 | 24/41 | 58.5 | 4/23 | 17.4 | [78] | |
| Lajeunie et al. (1999) | 36 | Cleft palate in 15 patients | 41.7 | 12/23 | 52.2 | 2/12 | 16.7 | One fetus with the S252F mutation also had a cleft palate | [17] |
| Sakai et al. (2001) | 6 | Cleft palate in 5 patients | 83.3 | 5/5 | 100.0 | 0/1 | 0.0 | [81] | |
| Von Gernet et al. (2000) | 21 | Cleft palate in 11 patients | 52.4 | 9/15 | 60.0 | 2/6 | 33.3 | [79] | |
| Kilcoyne et al. (2022) | 51 | Cleft palate or bifid uvula in 26 patients. | 51.0 | 18/28 | 64.3 | 8/23 | 34.8 | [80] | |
| Study | Model | Palatal Phenotypes | References |
|---|---|---|---|
| Wang et al. (2005) | Fgfr2+/S252W | Malformation of the palate. The structures of the face and palate were 40–50% smaller in the mutant mouse than in the control mouse. Incomplete closure of the anterior end of the secondary palate at P1. | [84] |
| Wang et al. (2010) | Fgfr2+/P253R | From E15.5 until P0, all Fgfr2+/P253R exhibited bilateral incomplete fusion at the junction of the primary and secondary palatal shelves. The developing mutant palates were shorter. | [85] |
| Martínez-Abadías et al. (2013) | Fgfr2+/S252W and Fgfr2+/P253R | Contracted and separated palatal shelves, and premature fusion of the maxillary–palatine suture in Fgfr2+/S252W and Fgfr2+/P253R mice. Fgfr2+/S252W mice display relatively more severe palatal dysmorphology. | [91] |
| Holmes; Basilico (2012) | EIIA-Cre;Fgfr2NeoS252W/+ | Incomplete closure of the anterior end of the secondary palate at P0. | [90] |
| Wnt1-Cre;Fgfr2NeoS252W/+ | Complete palatal closure at P0. | [90] | |
| Mesp1-Cre;Fgfr2NeoS252W/+ | Complete palatal closure at P0. | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willie, D.; Holmes, G.; Jabs, E.W.; Wu, M. Cleft Palate in Apert Syndrome. J. Dev. Biol. 2022, 10, 33. https://doi.org/10.3390/jdb10030033
Willie D, Holmes G, Jabs EW, Wu M. Cleft Palate in Apert Syndrome. Journal of Developmental Biology. 2022; 10(3):33. https://doi.org/10.3390/jdb10030033
Chicago/Turabian StyleWillie, Delayna, Greg Holmes, Ethylin Wang Jabs, and Meng Wu. 2022. "Cleft Palate in Apert Syndrome" Journal of Developmental Biology 10, no. 3: 33. https://doi.org/10.3390/jdb10030033
APA StyleWillie, D., Holmes, G., Jabs, E. W., & Wu, M. (2022). Cleft Palate in Apert Syndrome. Journal of Developmental Biology, 10(3), 33. https://doi.org/10.3390/jdb10030033