Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology



{kind=link}
Abstract
1. Introduction
2. The Discovery of NET Formation
3. The Role of NET Formation in Complement Activation
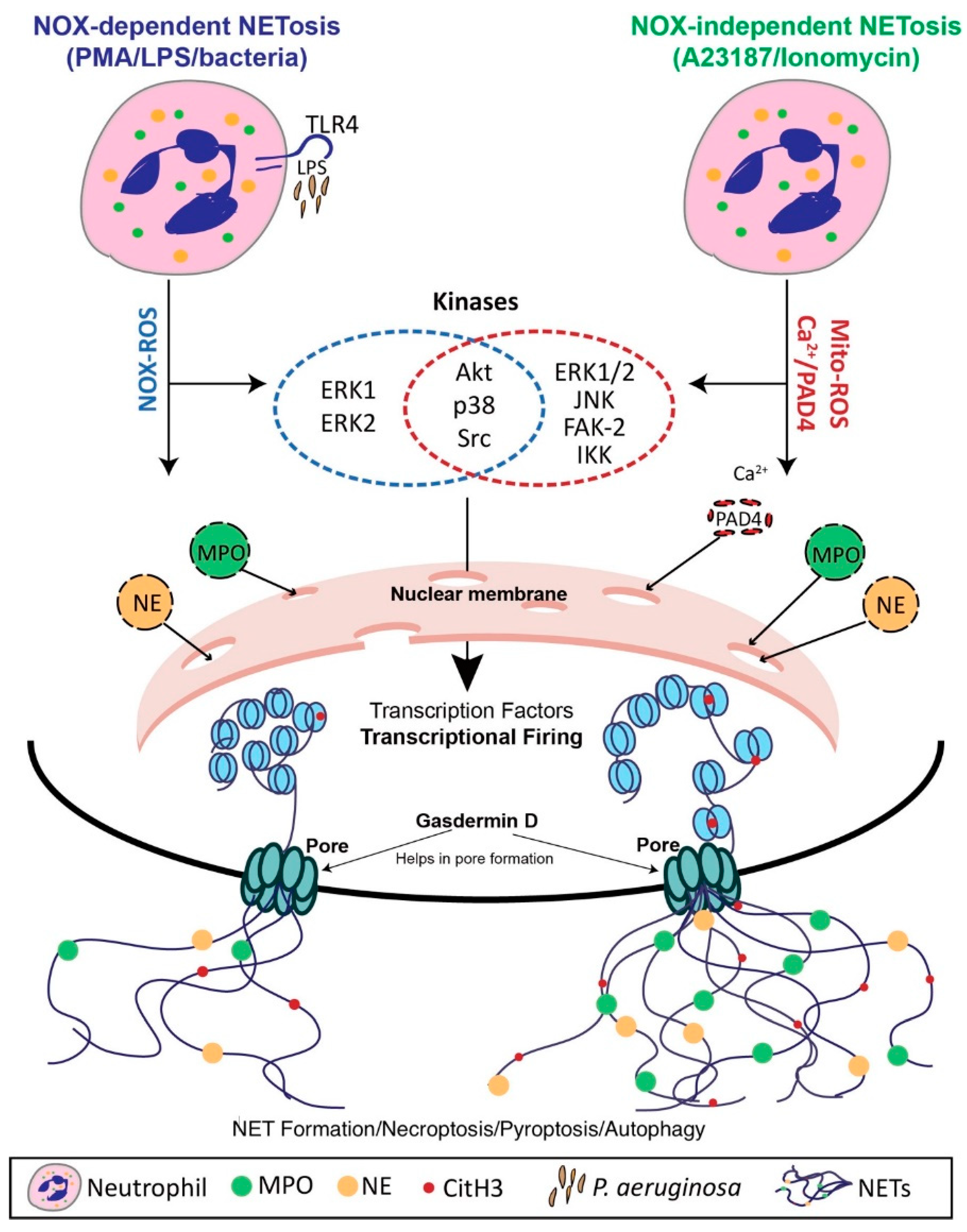
4. Mechanisms of NET Formation
4.1. Nox-Dependent NET Formation
4.2. Nox-Independent NET Formation
4.3. The Role of Transcriptional Firing in NET Formation
4.4. Vital NET Formation
4.5. The Role of Histone Modifications in NET Formation
5. NET Formation in Various Physiological Contexts
6. Clearance of Extruded NETs
7. NET-Induced Pathology
7.1. Immunodeficiency to Autoimmunity
7.2. Diabetes and Cardiovascular Disease
7.3. Cancer and Cystic Fibrosis
8. Potential Pharmacological Manipulations of NET Formation and Extruded NETs
8.1. Manipulation of NET Formation
8.2. Manipulation of Extruded NETs
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Olga Tatsiy, P.P.M. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 484. [Google Scholar] [CrossRef] [PubMed]
- Naffah de Souza, C.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Câmara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front. Immunol. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Nadesalingam, A.; Chen, J.H.K.; Farahvash, A.; Khan, M.A. Hypertonic Saline Suppresses NADPH Oxidase-Dependent Neutrophil Extracellular Trap Formation and Promotes Apoptosis. Front. Immunol. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 8, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yousefi, S.; Simon, H.-U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 2018, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Yoo, D.-G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of Cystic Fibrosis Airway Inflammatory Markers from Pseudomonas aeruginosa–Stimulated Human Neutrophils Involves NADPH Oxidase-Dependent Extracellular DNA Trap Formation. J. Immunol. 2014, 192, 4728–4738. [Google Scholar] [CrossRef]
- Douda, D.N.; Yip, L.; Khan, M.A.; Grasemann, H.; Palaniyar, N. Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood 2014, 123, 597–600. [Google Scholar] [CrossRef]
- Keshari, R.S.; Verma, A.; Barthwal, M.K.; Dikshit, M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J. Cell. Biochem. 2013, 114, 532–540. [Google Scholar] [CrossRef]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Malawista, S.E.; De Boisfleury Chevance, A. The cytokineplast: Purified, stable, and functional motile machinery from human blood polymorphonuclear leukocytes. J. Cell Biol. 1982, 95, 960–973. [Google Scholar] [CrossRef]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Neeli, I.; Khan, S.N.; Radic, M. Histone Deimination as a Response to Inflammatory Stimuli in Neutrophils. J. Immunol. 2008, 180, 1895–1902. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 2012, 120, 3118–3125. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Azzouz, D.; Khan, M.A.; Sweezey, N.; Palaniyar, N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137. [Google Scholar] [CrossRef]
- Jose, P.J. Complement-derived peptide mediators of inflammation. Br. Med. Bull. 1987, 43, 336–349. [Google Scholar] [CrossRef]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.-H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2018, 16, 19. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.-C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Fay, A.J.; Qian, X.; Jan, Y.N.; Jan, L.Y. SK channels mediate NADPH oxidase-independent reactive oxygen species production and apoptosis in granulocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 17548–17553. [Google Scholar] [CrossRef]
- Borregaard, N.; Sørensen, O.E.; Theilgaard-Mönch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef]
- Subrahmanyam, Y.V.B.K.; Yamaga, S.; Prashar, Y.; Lee, H.H.; Hoe, N.P.; Kluger, Y.; Gerstein, M.; Goguen, J.D.; Newburger, P.E.; Weissman, S.M. RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 2001, 97, 2457–2468. [Google Scholar] [CrossRef]
- Sollberger, G.; Amulic, B.; Zychlinsky, A. Neutrophil Extracellular Trap Formation Is Independent of De Novo Gene Expression. PLoS ONE 2016, 11, e0157454. [Google Scholar] [CrossRef]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U.; et al. NADPH Oxidase–Independent Formation of Extracellular DNA Traps by Basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef]
- Wang, Y. Human PAD4 Regulates Histone Arginine Methylation Levels via Demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef]
- Raijmakers, R.; Zendman, A.J.W.; Egberts, W.V.; Vossenaar, E.R.; Raats, J.; Soede-Huijbregts, C.; Rutjes, F.P.J.T.; van Veelen, P.A.; Drijfhout, J.W.; Pruijn, G.J.M. Methylation of arginine residues interferes with citrullination by peptidylarginine deiminases in vitro. J. Mol. Biol. 2007, 367, 1118–1129. [Google Scholar] [CrossRef]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J. Inflamm. 2010, 7, 9. [Google Scholar] [CrossRef]
- Hamam, H.J.; Palaniyar, N. Histone Deacetylase Inhibitors Dose-Dependently Switch Neutrophil Death from NETosis to Apoptosis. Biomolecules 2019, 9, 184. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Grasemann, H.; Sweezey, N.; Palaniyar, N. Regulating NETosis: Increasing pH Promotes NADPH Oxidase-Dependent NETosis. Front. Med. 2018, 5, 19. [Google Scholar] [CrossRef]
- Behnen, M.; Möller, S.; Brozek, A.; Klinger, M.; Laskay, T. Extracellular Acidification Inhibits the ROS-Dependent Formation of Neutrophil Extracellular Traps. Front. Immunol. 2017, 8, 184. [Google Scholar] [CrossRef]
- Maueröder, C.; Mahajan, A.; Paulus, S.; Gößwein, S.; Hahn, J.; Kienhöfer, D.; Biermann, M.H.; Tripal, P.; Friedrich, R.P.; Munoz, L.E.; et al. Ménage-à-Trois: The Ratio of Bicarbonate to CO2 and the pH Regulate the Capacity of Neutrophils to Form NETs. Front. Immunol. 2016, 7, 583. [Google Scholar] [CrossRef]
- Jones, E.M.; Cochrane, C.A.; Percival, S.L. The Effect of pH on the Extracellular Matrix and Biofilms. Adv. Wound Care 2015, 4, 431–439. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef]
- Korkmaz, H.I.; Ibrahim Korkmaz, H.; Ulrich, M.M.W.; Vogels, S.; de Wit, T.; van Zuijlen, P.P.M.; Krijnen, P.A.J.; Niessen, H.W.M. Neutrophil extracellular traps coincide with a pro-coagulant status of microcirculatory endothelium in burn wounds. Wound Repair Regen. 2017, 25, 609–617. [Google Scholar] [CrossRef]
- Vargas, A.; Boivin, R.; Cano, P.; Murcia, Y.; Bazin, I.; Lavoie, J.-P. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir. Res. 2017, 18, 207. [Google Scholar] [CrossRef]
- Farrera, C.; Fadeel, B. Macrophage Clearance of Neutrophil Extracellular Traps Is a Silent Process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef]
- Grégoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.-L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef]
- Wang, W.; Peng, W.; Ning, X. Increased levels of neutrophil extracellular trap remnants in the serum of patients with rheumatoid arthritis. Int. J. Rheum. Dis. 2018, 21, 415–421. [Google Scholar] [CrossRef]
- Papadaki, G.; Choulaki, C.; Mitroulis, I.; Verginis, P.; Repa, A.; Raptopoulou, A.; Boumpas, D.; Sidiropoulos, P. Enhanced release of neutrophil extracellular traps from peripheral blood neutrophils in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, A79. [Google Scholar] [CrossRef]
- Sur Chowdhury, C.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Ronald Kahn, C.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; ten Cate, H.; Hofstra, L.; et al. Elevated Levels of Circulating DNA and Chromatin Are Independently Associated with Severe Coronary Atherosclerosis and a Prothrombotic State. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef]
- Acuff, H.B.; Carter, K.J.; Fingleton, B.; Gorden, D.L.; Matrisian, L.M. Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. 2006, 66, 259–266. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kinsella, M.G.; Wight, T.N. Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am. J. Pathol. 1993, 143, 907–917. [Google Scholar]
- Ostafin, M.; Pruchniak, M.P.; Ciepiela, O.; Reznick, A.Z.; Demkow, U. Different procedures of diphenyleneiodonium chloride addition affect neutrophil extracellular trap formation. Anal. Biochem. 2016, 509, 60–66. [Google Scholar] [CrossRef]
- Rahman, S.; Gadjeva, M. Does NETosis Contribute to the Bacterial Pathoadaptation in Cystic Fibrosis? Front. Immunol. 2014, 5, 378. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Schön, M.P. Neutrophil extracellular traps: Protagonists of cancer progression? Oncogene 2017, 36, 2483–2490. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules 2019, 9, 365. https://doi.org/10.3390/biom9080365
Ravindran M, Khan MA, Palaniyar N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules. 2019; 9(8):365. https://doi.org/10.3390/biom9080365
Chicago/Turabian StyleRavindran, Mithunan, Meraj A. Khan, and Nades Palaniyar. 2019. "Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology" Biomolecules 9, no. 8: 365. https://doi.org/10.3390/biom9080365
APA StyleRavindran, M., Khan, M. A., & Palaniyar, N. (2019). Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules, 9(8), 365. https://doi.org/10.3390/biom9080365