Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Cell Counting Kit-8 (CCK-8) Assay
2.3. Hoechst 33258 Staining
2.4. Colony Formation Assay
2.5. Cell Cycle Analysis
2.6. Annexin V-FITC/Propidium Iodide (PI) Double Staining Assay
2.7. Mitochondrial Membrane Potential Assay
2.8. Reactive Oxygen Species Assay
2.9. Western Blotting Analysis
2.10. Statistical Analysis
3. Results
3.1. In Vitro Antigastric Cancer Effects of CBD
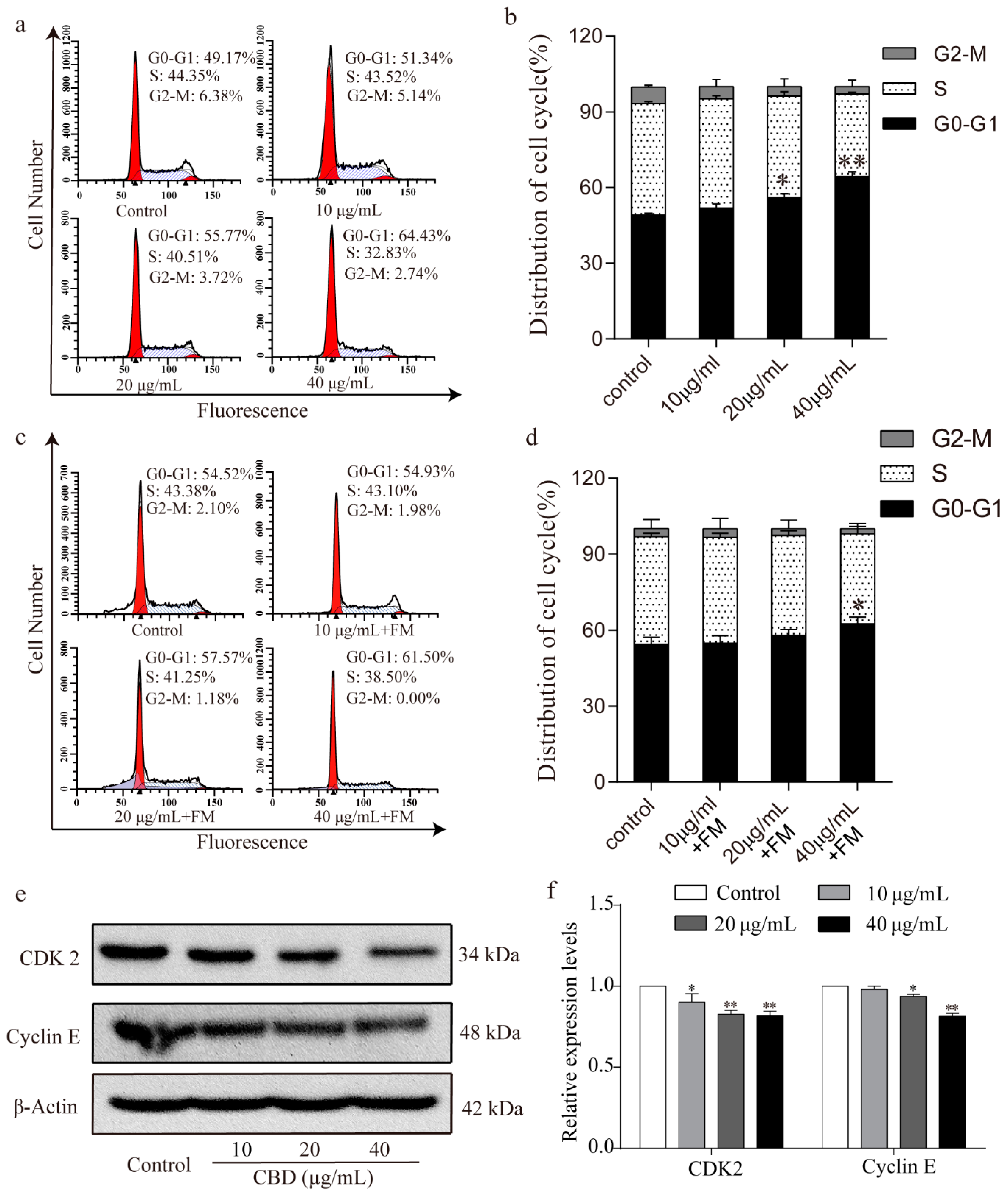
3.2. CBD Induces Cell Cycle Arrest at the G0–G1 Phase in SGC-7901 Cells
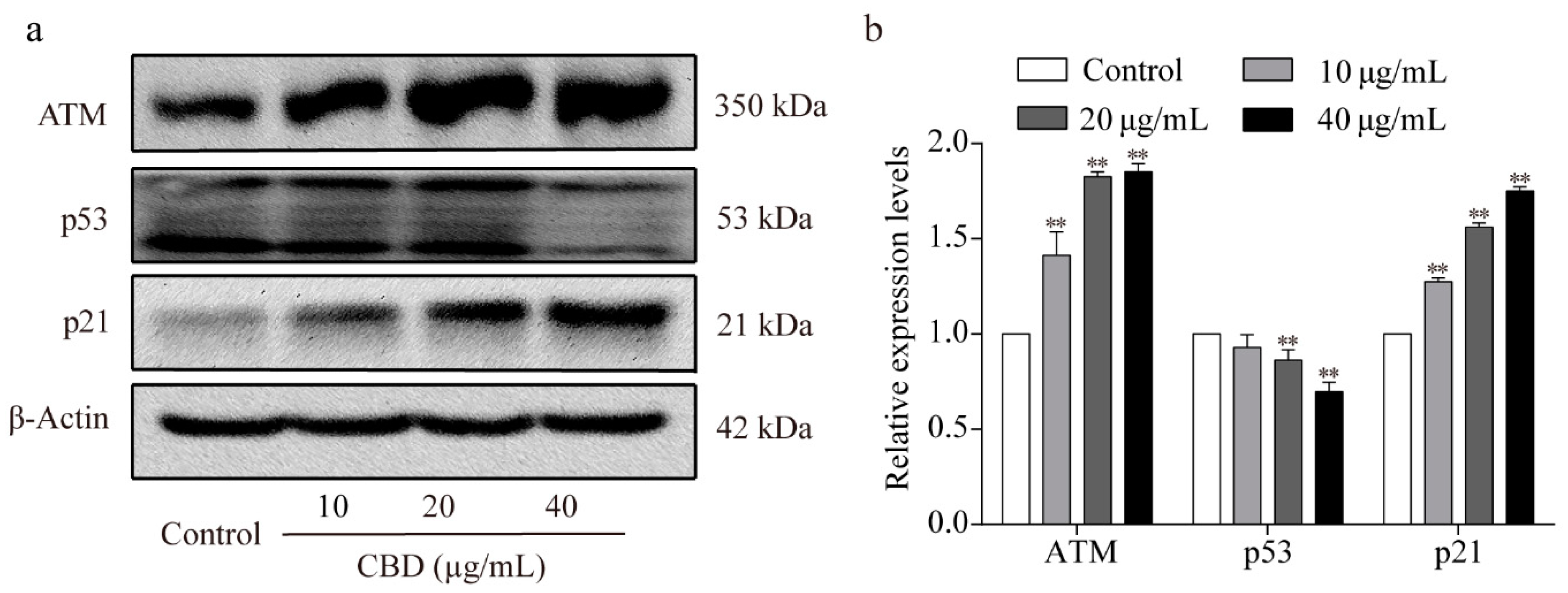
3.3. Effects of CBD on the ATM/p53/p21 Signaling Pathway
3.4. CBD Promotes Apoptosis in SGC-7901 Cells
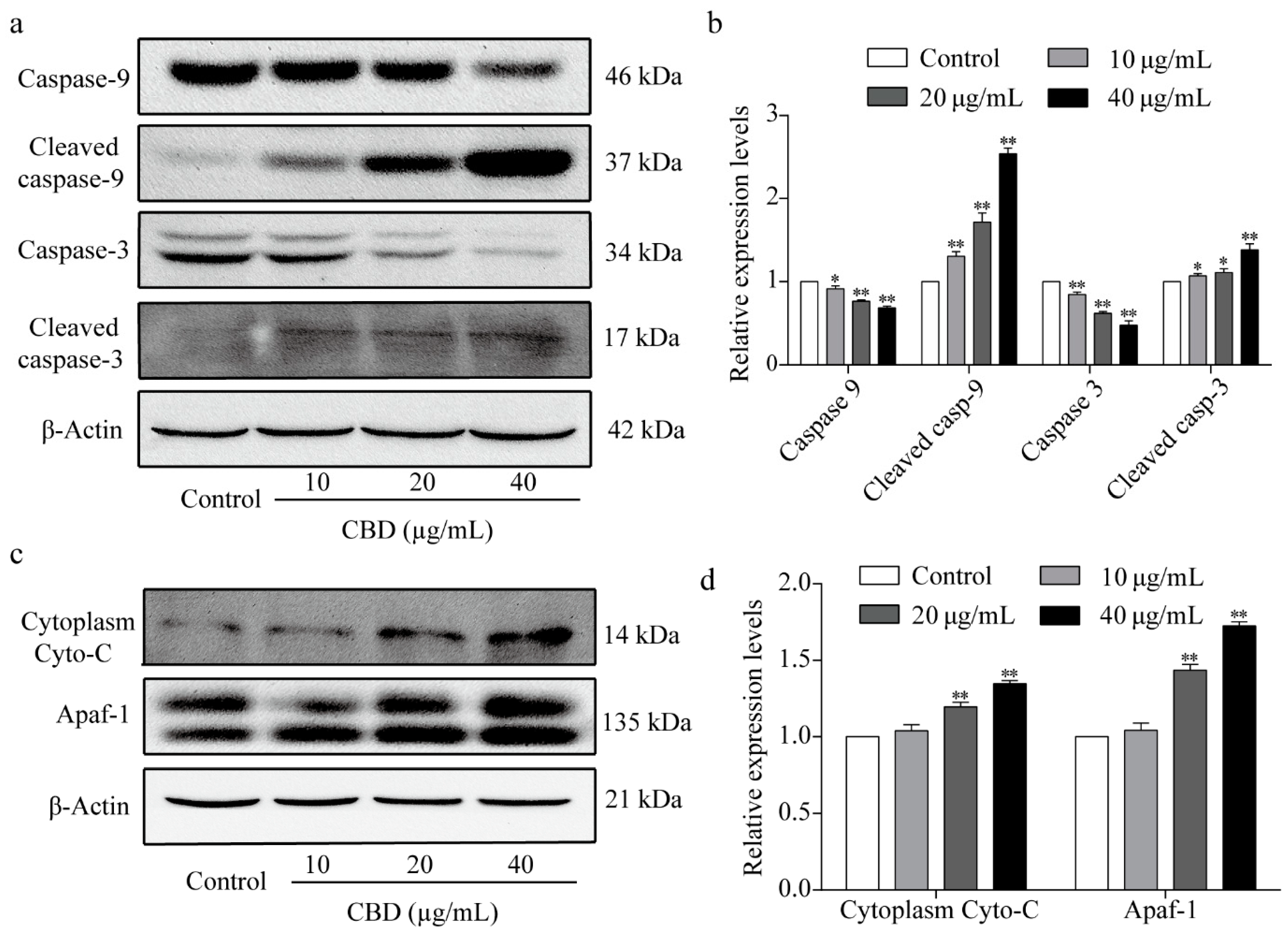
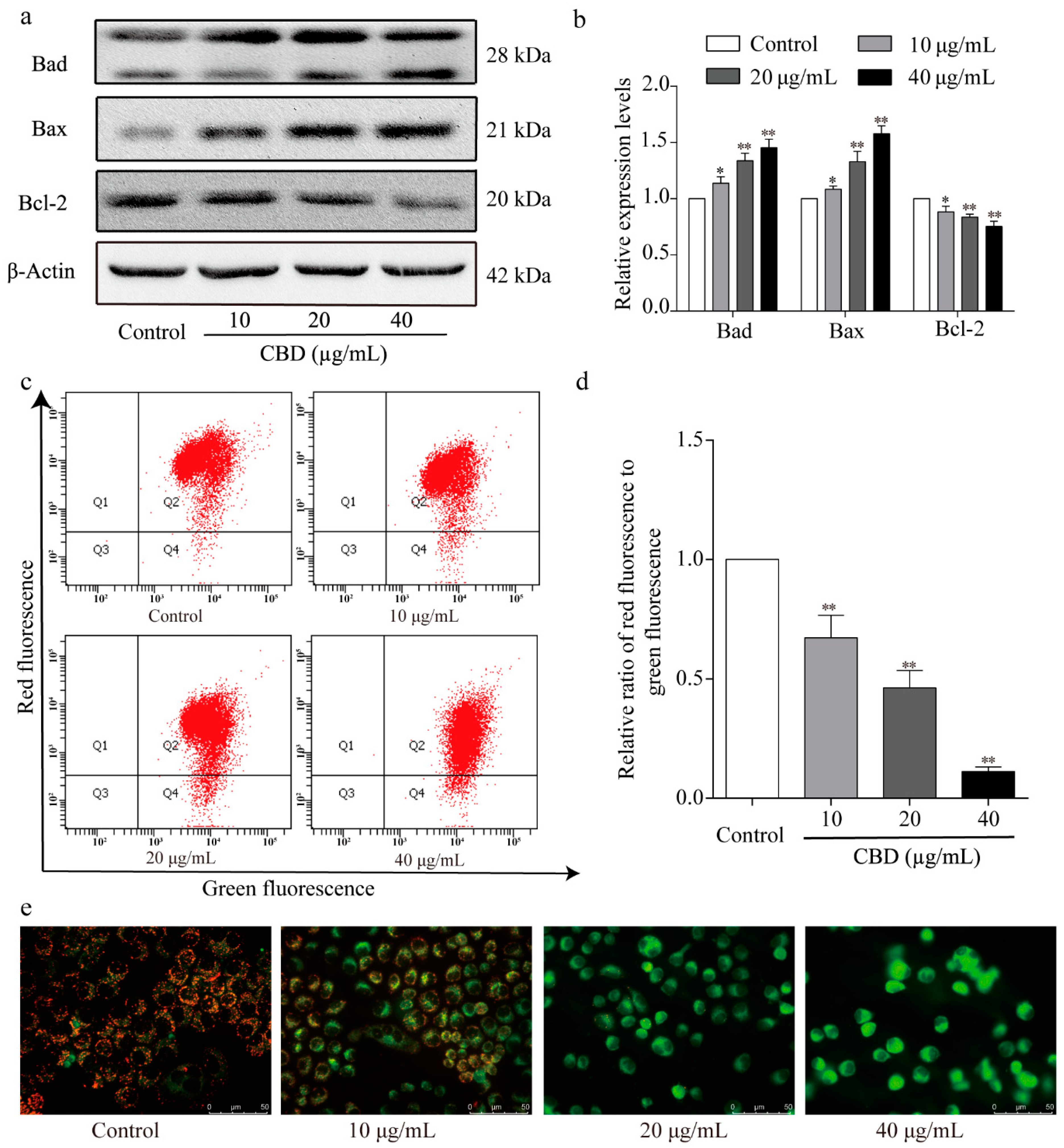
3.5. Effect of CBD on Apoptosis-Related Proteins
3.6. The Effect of CBD on the Mitochondrial Apoptosis Signaling Pathway
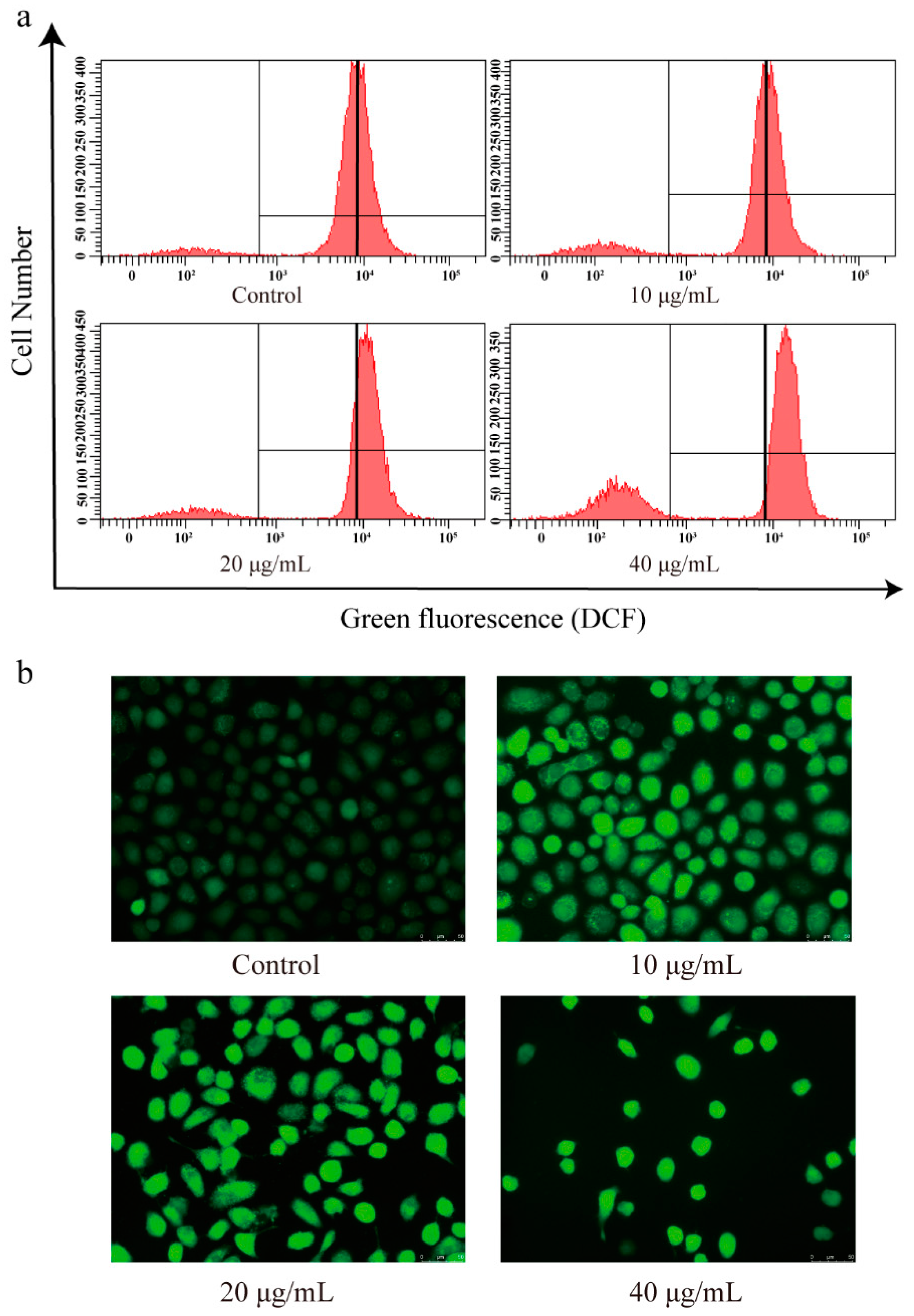
3.7. The Effect of CBD on the ROS Levels of SGC-7901 Cells
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Wang, G.; Yang, B.; Fu, Z.; Wang, X.; Zhang, Z. Efficacy and safety of oxaliplatin-based regimen versus cisplatin-based regimen in the treatment of gastric cancer: A meta-analysis of randomized controlled trials. Int. J. Clin. Oncol. 2019, 24, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, R.; Ren, G.; Zhao, J.; Li, H.; Guo, C.; He, W.; Wu, X.; Zhang, W. Differences in clinicopathological characteristics and computed tomography findings between signet ring cell carcinoma and nonsignet ring cell carcinoma in early and advanced gastric cancer. Cancer Med. 2018, 7, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Wang, C.C.; Lee, H.L.; Peng, C.M.; Yang, T.W.; Chen, H.Y.; Sung, W.W.; Lin, C.C. Health disparities are associated with gastric cancer mortality-to-incidence ratios in 57 countries. World J. Gastroenterol. 2017, 23, 7881–7887. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Q.T.; Liu, Y.P.; Dong, Q.Q.; Hu, H.J.; Miao, Z.; Li, S.; Liu, Y.; Zhou, H.; Zhang, T.C.; et al. ATM Signaling Pathway Is Implicated in the SMYD3-mediated Proliferation and Migration of Gastric Cancer Cells. J. Gastric. Cancer 2017, 17, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zhu, C.; Yuan, Y.; Feng, Q.; Feng, Y.; Hao, Y.; Li, J.; Zhang, K.; Ye, G.; Ye, L.; et al. Development and validation of a prediction rule for estimating gastric cancer risk in the Chinese high-risk population: A nationwide multicentre study. Gut 2019. [Google Scholar] [CrossRef]
- Liu, J.; Li, D.; Dang, L.; Liang, C.; Guo, B.; Lu, C.; He, X.; Cheung, H.Y.; He, B.; Liu, B.; et al. Osteoclastic miR-214 targets TRAF3 to contribute to osteolytic bone metastasis of breast cancer. Sci. Rep. 2017, 7, 40487. [Google Scholar] [CrossRef]
- He, Y.; Mao, M.; Shi, W.; He, Z.; Zhang, L.; Wang, X. Development and validation of a prognostic nomogram in gastric cancer with hepatitis B virus infection. J. Transl. Med. 2019, 17, 98. [Google Scholar] [CrossRef]
- Kiyokawa, T.; Fukagawa, T. Recent trends from the results of clinical trials on gastric cancer surgery. Cancer Commun. 2019, 39, 11. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Wang, Q.; Li, J.; Bai, B.; Li, Z.; Wu, X.; Yu, P.; Li, X.; Yin, J. Postoperative complications and prognosis after radical gastrectomy for gastric cancer: A systematic review and meta-analysis of observational studies. World J. Surg. Oncol. 2019, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Biagioni, A.; Cianchi, F.; Skalamera, I.; Staderini, F.; Schiavone, N.; Papucci, L.; Magnelli, L. Chemotherapy resistance-associated epithelial to endothelial transition in gastric cancer. J. Biol. Regul. Homeost. Agents 2018, 32 (Suppl. 1), 30. [Google Scholar] [PubMed]
- Liang Ong, S.C.; Batumaly, S.K.; Jusoh, S.M. Portal vein tumor thrombus from gastric cancer. J. Ultrason. 2018, 18, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Sawa, H.; Kobayashi, T.; Mukai, K.; Zhang, W.; Shiku, H. Bax overexpression enhances cytochrome c release from mitochondria and sensitizes KATOIII gastric cancer cells to chemotherapeutic agent-induced apoptosis. Int. J. Oncol. 2000, 16, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.B.; Jian, T.; Yue, C.; Chen, D.; Chen, W.; Bao, T.T.; Liu, H.X.; Cao, Y.; Li, W.B.; Yang, Z.; et al. Chemo-resistant Gastric Cancer Associated Gene Expression Signature: Bioinformatics Analysis Based on Gene Expression Omnibus. Anticancer Res. 2019, 39, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Guo, Z.; Zhu, P.; Chen, J.; Huang, Y. Traditional Chinese medicine as a cancer treatment: Modern perspectives of ancient but advanced science. Cancer Med. 2019, 8, 1958–1975. [Google Scholar] [CrossRef] [PubMed]
- Farrimond, J.A.; Whalley, B.J.; Williams, C.M. Cannabinol and cannabidiol exert opposing effects on rat feeding patterns. Psychopharmacology (Berl) 2012, 223, 117–129. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E.; Sun, Y.; Bennett, A.J.; Randall, M.D.; Kendall, D.A. Time-dependent vascular actions of cannabidiol in the rat aorta. Eur. J. Pharmacol. 2009, 612, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Hinz, B. Cannabinoids as Anticancer Drugs. Adv. Pharmacol. 2017, 80, 397–436. [Google Scholar]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty acid-binding proteins (FABPs) are intracellular carriers for Delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef]
- Jacobsson, S.O.; Rongard, E.; Stridh, M.; Tiger, G.; Fowler, C.J. Serum-dependent effects of tamoxifen and cannabinoids upon C6 glioma cell viability. Biochem. Pharmacol. 2000, 60, 1807–1813. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Ligresti, A.; Schiano Moriello, A.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2013, 168, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- Bergamaschi, M.M.; Queiroz, R.H.; Zuardi, A.W.; Crippa, J.A. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 2011, 6, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, D.; Faraone, I.; Laurenzana, I.; Milella, L.; Trino, S.; De Luca, L.; Del Vecchio, L.; Armentano, M.F.; Sinisgalli, C.; Chiummiento, L.; et al. Future in the Past: Azorella glabra Wedd. as a Source of New Natural Compounds with Antiproliferative and Cytotoxic Activity on Multiple Myeloma Cells. Int. J. Mol. Sci. 2018, 19, 3348. [Google Scholar] [CrossRef]
- Lv, G.; Sun, D.; Zhang, J.; Xie, X.; Wu, X.; Fang, W.; Tian, J.; Yan, C.; Wang, H.; Fu, F. Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm. Sin. B 2017, 7, 52–58. [Google Scholar] [CrossRef]
- Yang, Y.; Guan, D.; Lei, L.; Lu, J.; Liu, J.Q.; Yang, G.; Yan, C.; Zhai, R.; Tian, J.; Bi, Y.; et al. A novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol. Appl. Pharmacol. 2018, 341, 98–105. [Google Scholar] [CrossRef]
- Barta, T.; Vinarsky, V.; Holubcova, Z.; Dolezalova, D.; Verner, J.; Pospisilova, S.; Dvorak, P.; Hampl, A. Human embryonic stem cells are capable of executing G1/S checkpoint activation. Stem Cells 2010, 28, 1143–1152. [Google Scholar] [CrossRef]
- Delia, D.; Fontanella, E.; Ferrario, C.; Chessa, L.; Mizutani, S. DNA damage-induced cell-cycle phase regulation of p53 and p21waf1 in normal and ATM-defective cells. Oncogene 2003, 22, 7866–7869. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, Q.; Cui, H.; Deng, H.; Kuang, P.; Lu, Y.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; et al. Sodium fluoride causes hepatocellular S-phase arrest by activating ATM-p53-p21 and ATR-Chk1-Cdc25A pathways in mice. Oncotarget 2018, 9, 4318–4337. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Singh, S.P.; Hader, D.P.; Sinha, R.P. Detection of reactive oxygen species (ROS) by the oxidant-sensing probe 2′,7′-dichlorodihydrofluorescein diacetate in the cyanobacterium Anabaena variabilis PCC 7937. Biochem. Biophys. Res. Commun. 2010, 397, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Hausman-Kedem, M.; Menascu, S.; Kramer, U. Efficacy of CBD-enriched medical cannabis for treatment of refractory epilepsy in children and adolescents - An observational, longitudinal study. Brain Dev. 2018, 40, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Daris, B.; Tancer Verboten, M.; Knez, Z.; Ferk, P. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn. J. Basic Med. Sci. 2019, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulam, R.; Alvarez, L.; Guzman, M.; Galve-Roperh, I. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef]
- Gallily, R.; Even-Chena, T.; Katzavian, G.; Lehmann, D.; Dagan, A.; Mechoulam, R. Gamma-irradiation enhances apoptosis induced by cannabidiol, a non-psychotropic cannabinoid, in cultured HL-60 myeloblastic leukemia cells. Leuk. Lymphoma 2003, 44, 1767–1773. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) Is a Novel Inhibitor for Exosome and Microvesicle (EMV) Release in Cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Perez-Gomez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; Garcia-Real, I.; Palacios, J.; Manes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef]
- Zhong, J.; Chen, S.; Xue, M.; Du, Q.; Cai, J.; Jin, H.; Si, J.; Wang, L. ZIC1 modulates cell-cycle distributions and cell migration through regulation of sonic hedgehog, PI(3)K and MAPK signaling pathways in gastric cancer. BMC Cancer 2012, 12, 290. [Google Scholar] [CrossRef]
- Zang, Y.Q.; Feng, Y.Y.; Luo, Y.H.; Zhai, Y.Q.; Ju, X.Y.; Feng, Y.C.; Wang, J.R.; Yu, C.Q.; Jin, C.H. Glycitein induces reactive oxygen species-dependent apoptosis and G0/G1 cell cycle arrest through the MAPK/STAT3/NF-kappaB pathway in human gastric cancer cells. Drug Dev. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lan, B.; Liu, B.Y.; Cheng, X.H.; Zhang, J.; Wang, K.K.; Zhu, Z.G. [Analysis of gene expression profiles in gastric cancer cell cycle]. Zhonghua Zhong Liu Za Zhi 2006, 28, 568–571. [Google Scholar] [PubMed]
- Tan, H.; Gao, S.; Zhuang, Y.; Dong, Y.; Guan, W.; Zhang, K.; Xu, J.; Cui, J. R-Phycoerythrin Induces SGC-7901 Apoptosis by Arresting Cell Cycle at S Phase. Mar. Drugs 2016, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Barshishat, S.; Elgrably-Weiss, M.; Edelstein, J.; Georg, J.; Govindarajan, S.; Haviv, M.; Wright, P.R.; Hess, W.R.; Altuvia, S. OxyS small RNA induces cell cycle arrest to allow DNA damage repair. EMBO J. 2018, 37, 413–426. [Google Scholar] [CrossRef]
- Subhash, V.V.; Tan, S.H.; Yeo, M.S.; Yan, F.L.; Peethala, P.C.; Liem, N.; Krishnan, V.; Yong, W.P. ATM Expression Predicts Veliparib and Irinotecan Sensitivity in Gastric Cancer by Mediating P53-Independent Regulation of Cell Cycle and Apoptosis. Mol. Cancer Ther. 2016, 15, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.C.; Wu, J.F.; Wang, D.B.; Qin, R.; Zhang, H. Relationship between the expression of human telomerase reverse transcriptase gene and cell cycle regulators in gastric cancer and its significance. World J. Gastroenterol. 2003, 9, 427–431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Kim, J.; Lee, K.J.; Kim, J.S.; Rho, J.G.; Shin, J.J.; Song, W.K.; Lee, E.K.; Egan, J.M.; Kim, W. Cannabinoids Regulate Bcl-2 and Cyclin D2 Expression in Pancreatic beta Cells. PLoS One 2016, 11, e0150981. [Google Scholar]
- Liu, M.; Luo, X.J.; Liao, F.; Lei, X.F.; Dong, W.G. Noscapine induces mitochondria-mediated apoptosis in gastric cancer cells in vitro and in vivo. Cancer Chemother. Pharmacol. 2011, 67, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Vijayalakshmi, A.; Sindhu, G. Umbelliferone arrest cell cycle at G0/G1 phase and induces apoptosis in human oral carcinoma (KB) cells possibly via oxidative DNA damage. Biomed. Pharmacother. 2017, 92, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Almasi, S.; Long, C.Y.; Sterea, A.; Clements, D.R.; Gujar, S.; El Hiani, Y. TRPM2 Silencing Causes G2/M Arrest and Apoptosis in Lung Cancer Cells via Increasing Intracellular ROS and RNS Levels and Activating the JNK Pathway. Cell Physiol. Biochem. 2019, 52, 742–757. [Google Scholar] [PubMed]
- Chayapong, J.; Madhyastha, H.; Madhyastha, R.; Nurrahmah, Q.I.; Nakajima, Y.; Choijookhuu, N.; Hishikawa, Y.; Maruyama, M. Arsenic trioxide induces ROS activity and DNA damage, leading to G0/G1 extension in skin fibroblasts through the ATM-ATR-associated Chk pathway. Environ. Sci. Pollut. Res. Int. 2017, 24, 5316–5325. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Ma, Y.; Wang, J.; Yang, X.; Gu, Y.; Li, Y. In vitro and in vivo antitumor activity of neochlorogenic acid in human gastric carcinoma cells are complemented with ROS generation, loss of mitochondrial membrane potential and apoptosis induction. J. BUON 2019, 24, 221–226. [Google Scholar] [PubMed]
- Chung, Y.M.; Bae, Y.S.; Lee, S.Y. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic. Biol. Med. 2003, 34, 434–442. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules 2019, 9, 302. https://doi.org/10.3390/biom9080302
Zhang X, Qin Y, Pan Z, Li M, Liu X, Chen X, Qu G, Zhou L, Xu M, Zheng Q, et al. Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules. 2019; 9(8):302. https://doi.org/10.3390/biom9080302
Chicago/Turabian StyleZhang, Xin, Yao Qin, Zhaohai Pan, Minjing Li, Xiaona Liu, Xiaoyu Chen, Guiwu Qu, Ling Zhou, Maolei Xu, Qiusheng Zheng, and et al. 2019. "Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells" Biomolecules 9, no. 8: 302. https://doi.org/10.3390/biom9080302
APA StyleZhang, X., Qin, Y., Pan, Z., Li, M., Liu, X., Chen, X., Qu, G., Zhou, L., Xu, M., Zheng, Q., & Li, D. (2019). Cannabidiol Induces Cell Cycle Arrest and Cell Apoptosis in Human Gastric Cancer SGC-7901 Cells. Biomolecules, 9(8), 302. https://doi.org/10.3390/biom9080302