Dual Role of the Alternative Reading Frame ARF Protein in Cancer


Abstract
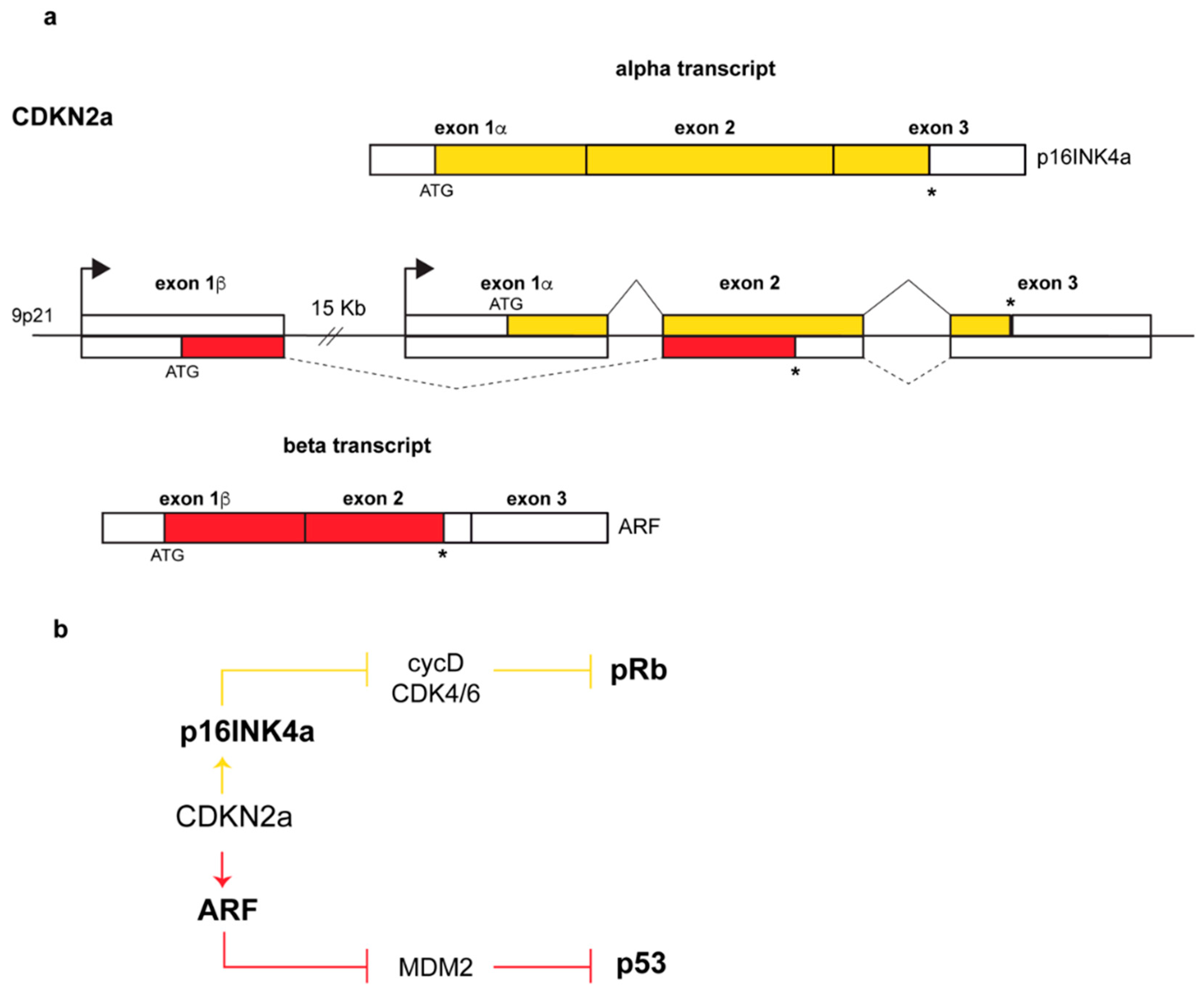
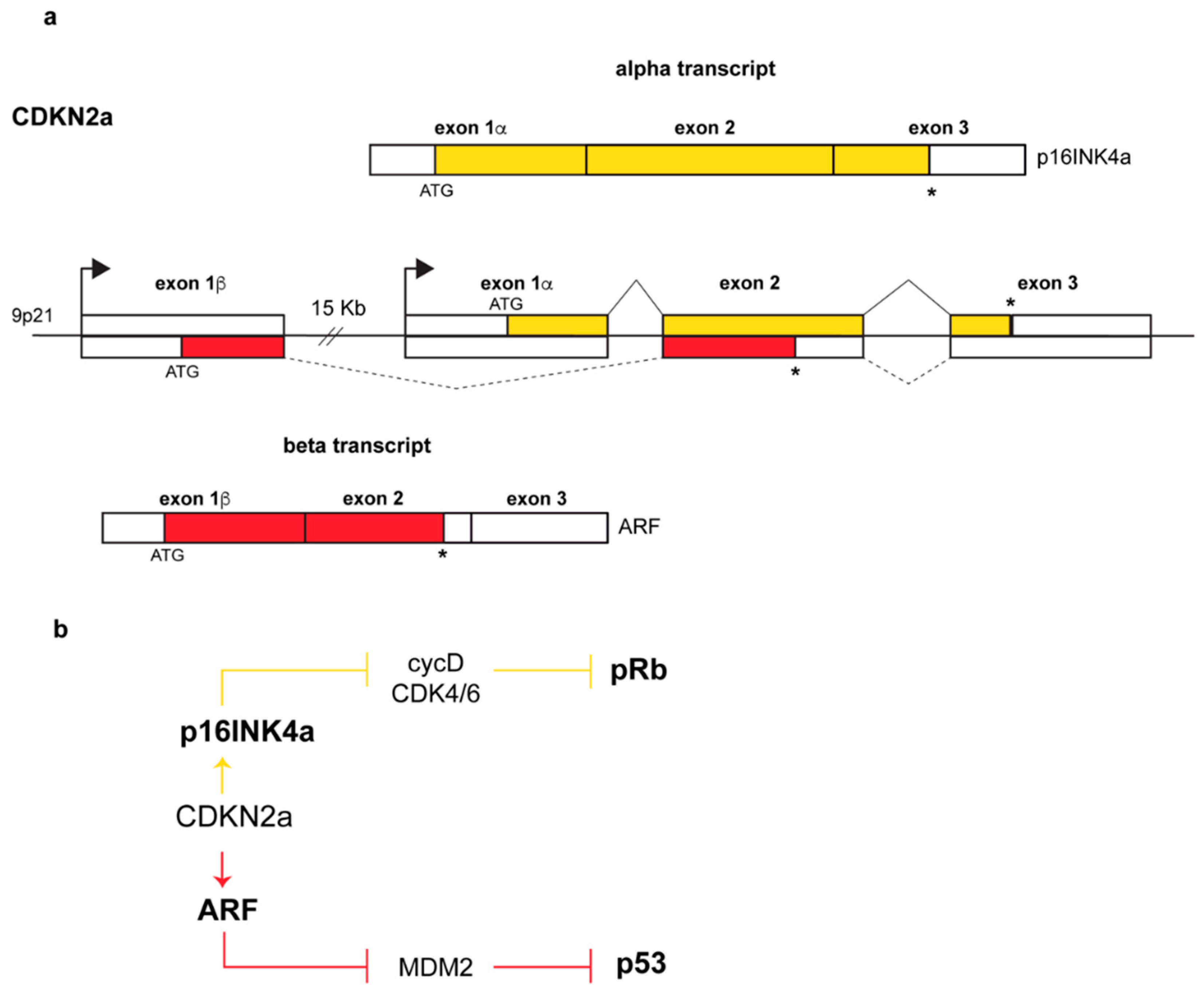
:1. Introduction
2. ARF Inactivation in Human and Murine Cancer
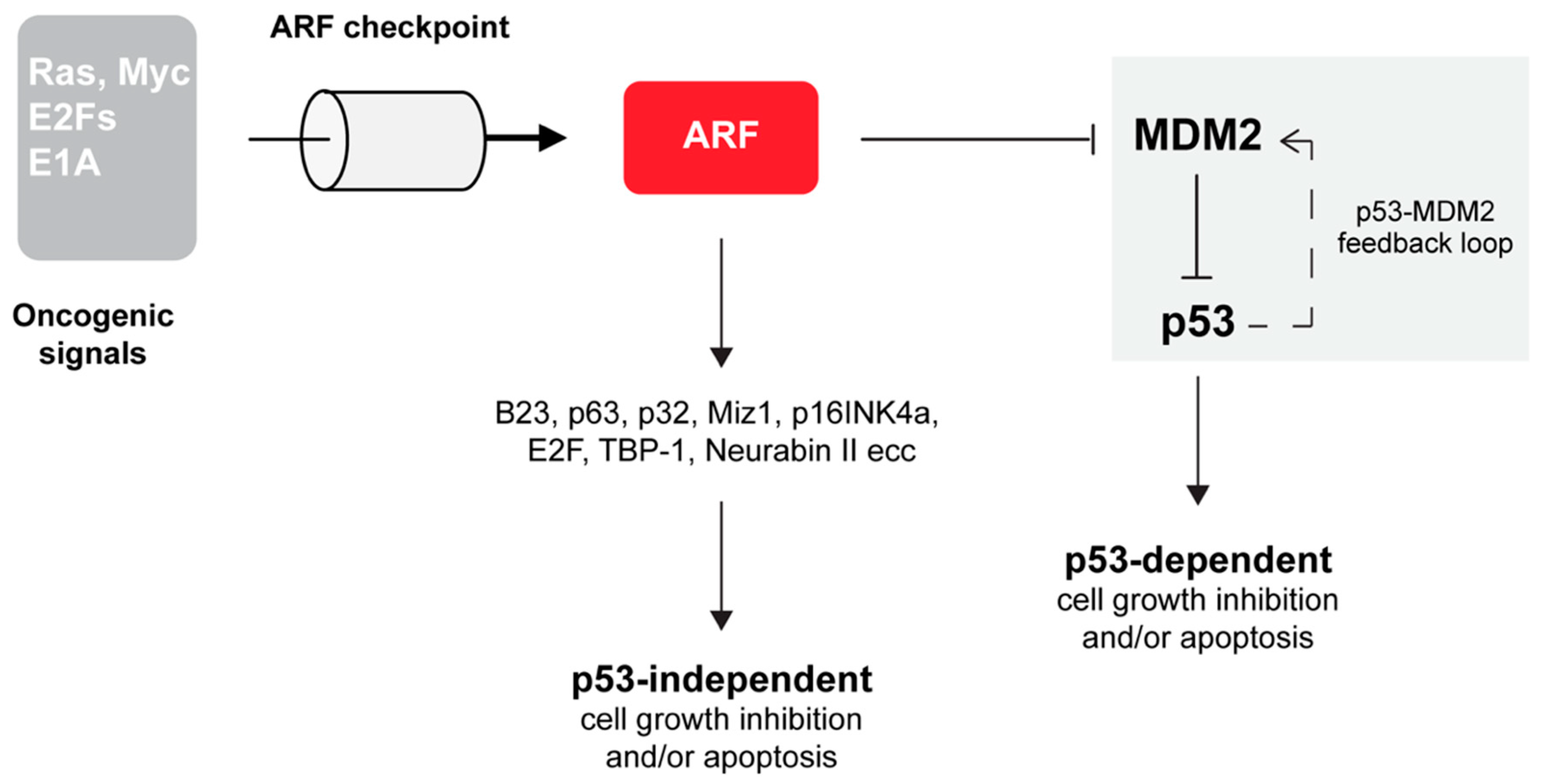
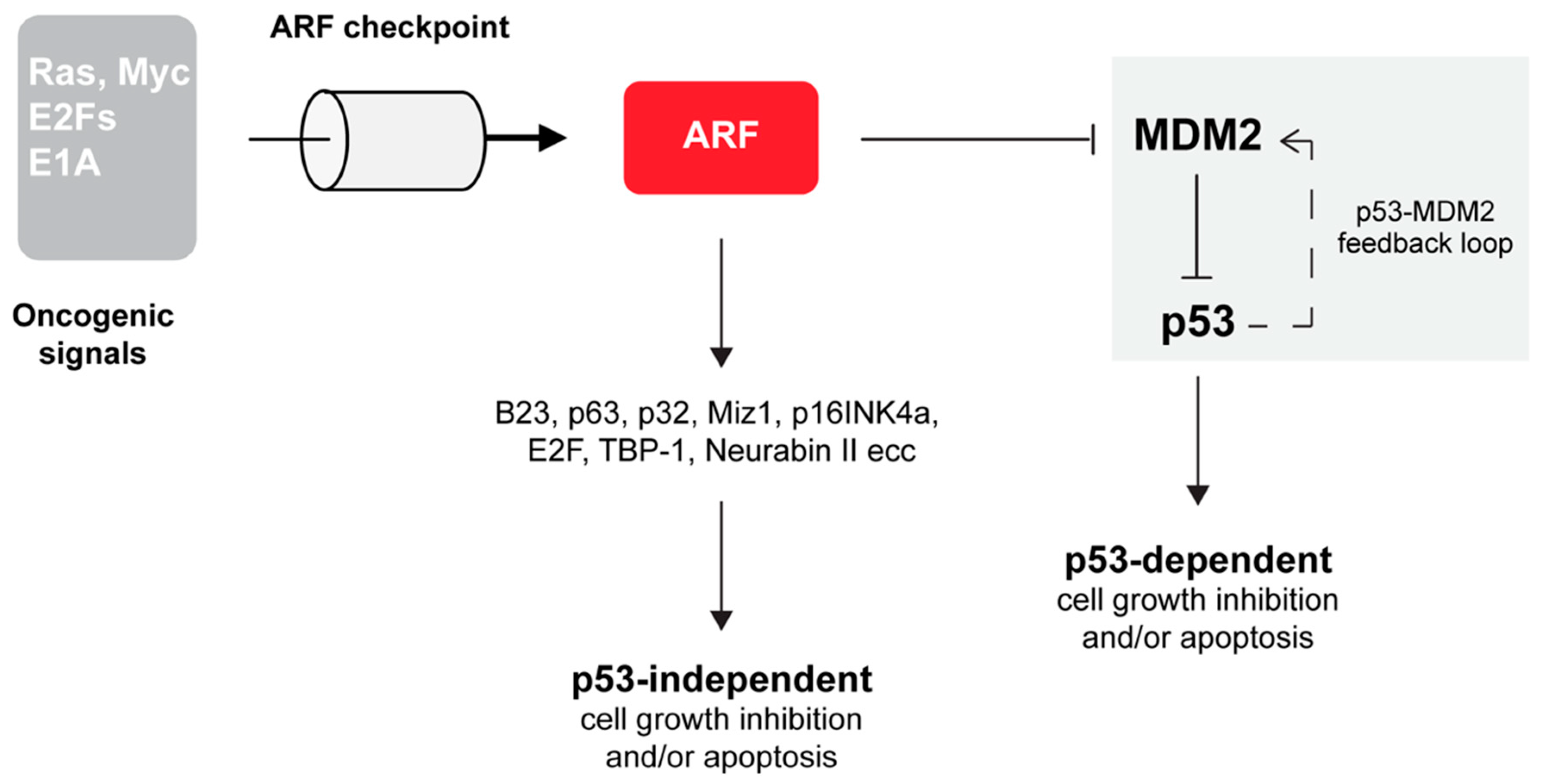
3. ARF is Involved in the p53 Pathway
ARF Inhibits MDM2 Through Multiple Mechanisms
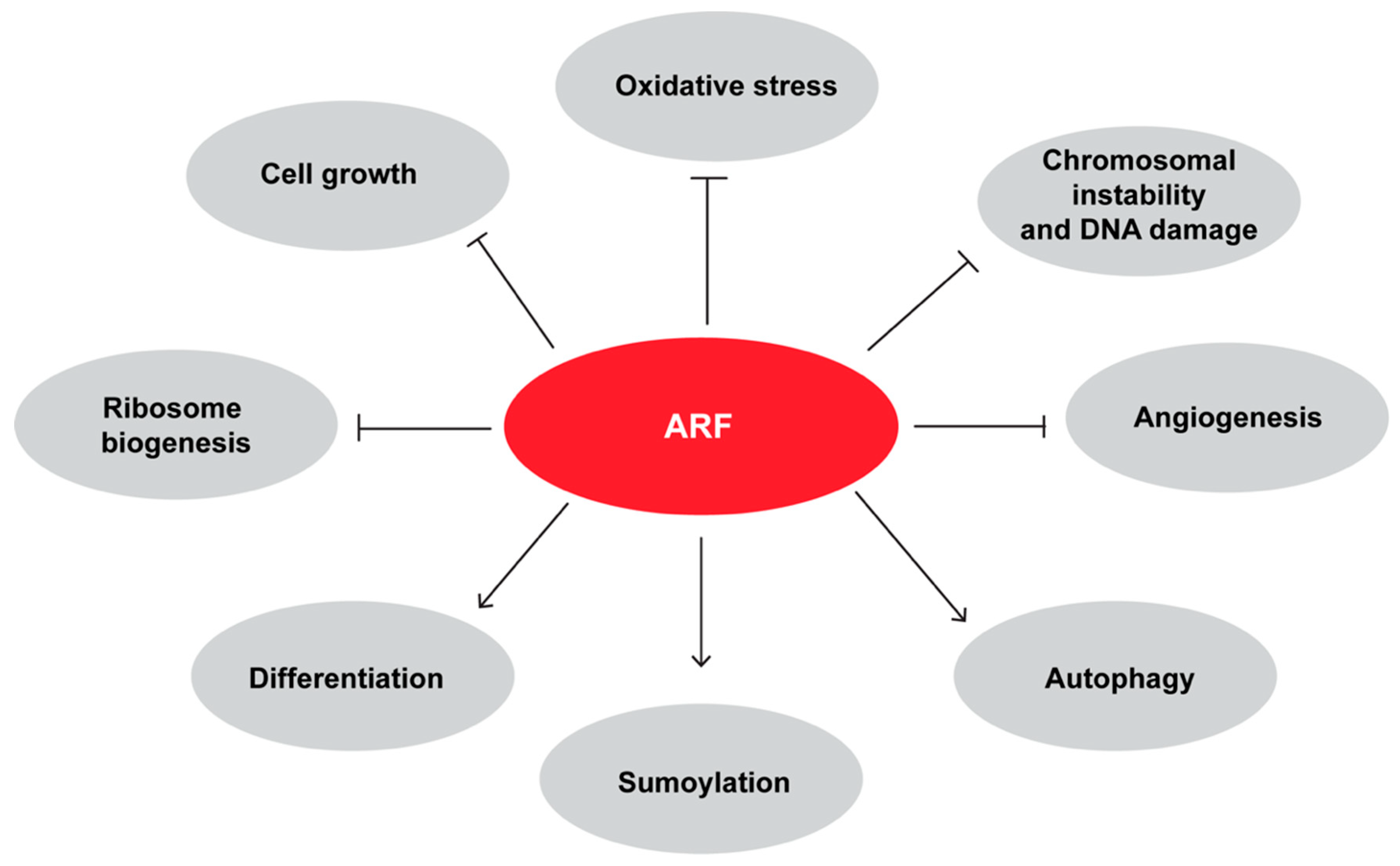
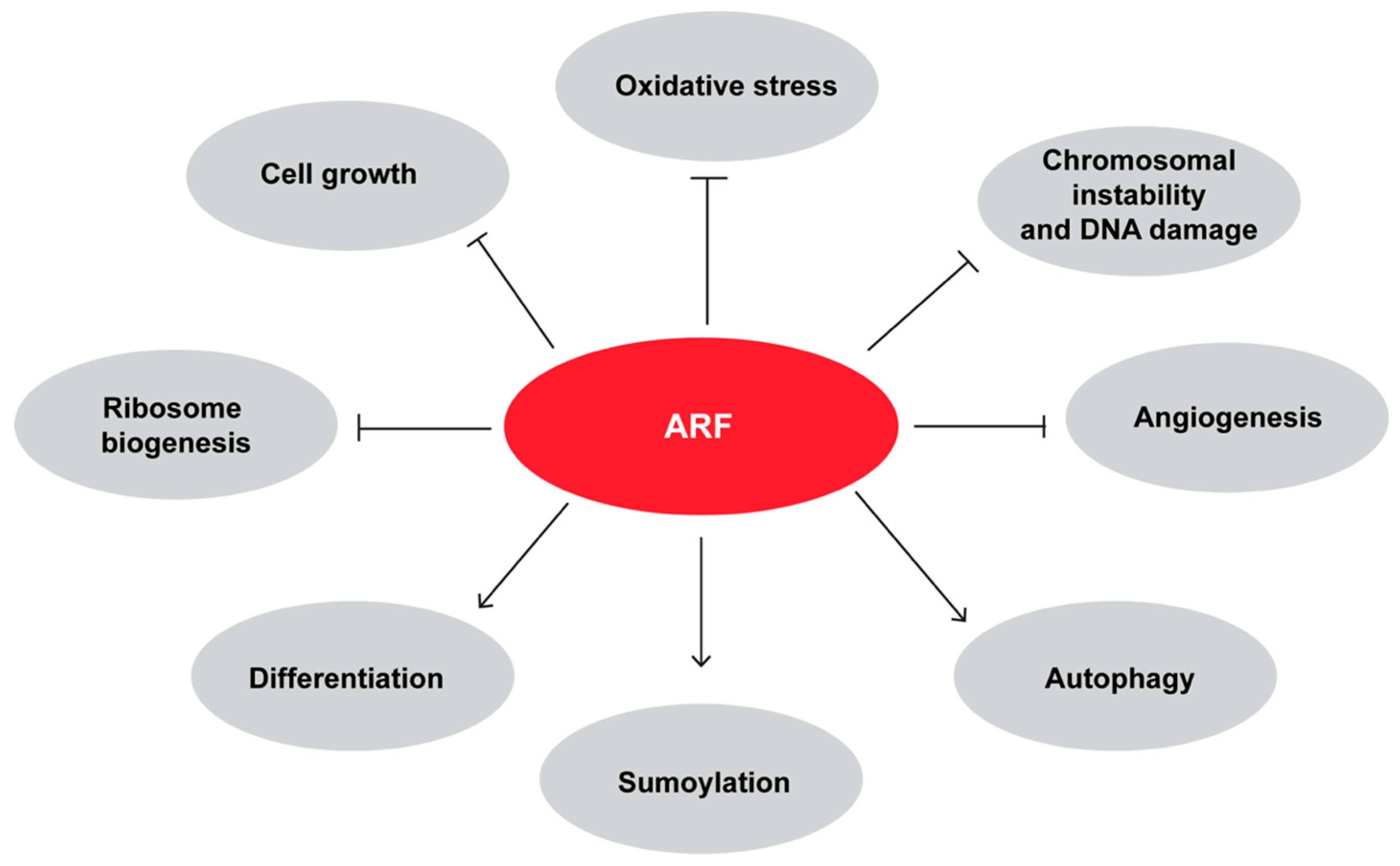
4. ARF is Involved In p53 Independent Pathways of Tumor Suppression
5. Mechanisms Governing ARF Degradation
6. ARF Plays a Role During Development and Differentiation
7. ARF has Pro-Oncogenic Functions
8. ARF and Autophagy
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Tommaso, A.; Hagen, J.; Tompkins, V.; Muniz, V.; Dudakovic, A.; Kitzis, A.; Ladeveze, V.; Quelle, D.E. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Exp. Cell Res. 2009, 315, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Soussi, T.; Shomer, B.; Hollstein, M.; Greenblatt, M.; Hovig, E.; Harris, C.C.; Montesano, R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997, 25, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and CDK inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2a locus, p14(Arf), participates in a regulatory feedback loop with p53 and mdm2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase CDK6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R.A. The INK4a/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the ink4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [PubMed]
- Sherr, C.J. Divorcing arf and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The Arf tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- De Stanchina, E.; McCurrach, M.E.; Zindy, F.; Shieh, S.Y.; Ferbeyre, G.; Samuelson, A.V.; Prives, C.; Roussel, M.F.; Sherr, C.J.; Lowe, S.W. E1a signaling to p53 involves the p19(Arf) tumor suppressor. Genes Dev. 1998, 12, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.P.; Barnes, J.M.; Paliwal, S.; Zhang, X.; Tang, X.; Chen, S.; Zamba, K.D.; Cullen, J.J.; Meyerholz, D.K.; Meyers, S.; et al. The ARF tumor suppressor inhibits tumor cell colonization independent of p53 in a novel mouse model of pancreatic ductal adenocarcinoma metastasis. Mol. Cancer Res. 2011, 9, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ishikawa, T.; Sugihara, E.; Kuninaka, S.; Miyamoto, T.; Mabuchi, Y.; Matsuzaki, Y.; Tsunoda, T.; Miya, F.; Morioka, H.; et al. c-Myc overexpression with loss of INK4a/ARF transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene 2010, 29, 5687–5699. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Bodner, S.; van de Kamp, E.; Randle, D.H.; Sherr, C.J. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999, 59, 2217–2222. [Google Scholar] [PubMed]
- Del Arroyo, A.G.; Peters, G. The INK4a/ARF network—Cell cycle checkpoint or emergency brake? Adv. Exp. Med. Biol. 2005, 570, 227–247. [Google Scholar] [PubMed]
- Rizos, H.; Darmanian, A.P.; Holland, E.A.; Mann, G.J.; Kefford, R.F. Mutations in the INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J. Biol. Chem. 2001, 276, 41424–41434. [Google Scholar] [CrossRef] [PubMed]
- Gardie, B.; Cayuela, J.M.; Martini, S.; Sigaux, F. Genomic alterations of the p19ARF encoding exons in T-cell acute lymphoblastic leukemia. Blood 1998, 91, 1016–1020. [Google Scholar] [PubMed]
- Rutter, J.L.; Goldstein, A.M.; Davila, M.R.; Tucker, M.A.; Struewing, J.P. CDKN2a point mutations d153spl(c.457g > t) and ivs 2 + 1g > t result in aberrant splice products affecting both p16ink4a and p14ARF. Oncogene 2003, 22, 4444–4448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Maggi, L.B., Jr.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar] [PubMed]
- Tsujimoto, H.; Hagiwara, A.; Sugihara, H.; Hattori, T.; Yamagishi, H. Promoter methylations of p16INK4a and p14Arf genes in early and advanced gastric cancer. Correlations of the modes of their occurrence with histologic type. Pathol. Res. Pract. 2002, 198, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Konishi, N.; Nakamura, M.; Kishi, M.; Nishimine, M.; Ishida, E.; Shimada, K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am. J. Pathol. 2002, 160, 1207–1214. [Google Scholar] [CrossRef]
- Hewitt, C.; Lee Wu, C.; Evans, G.; Howell, A.; Elles, R.G.; Jordan, R.; Sloan, P.; Read, A.P.; Thakker, N. Germline mutation of ARF in a melanoma kindred. Hum. Mol. Genet. 2002, 11, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Rizos, H.; Puig, S.; Badenas, C.; Malvehy, J.; Darmanian, A.P.; Jimenez, L.; Mila, M.; Kefford, R.F. A melanoma-associated germline mutation in exon 1β inactivates p14ARF. Oncogene 2001, 20, 5543–5547. [Google Scholar] [CrossRef] [PubMed]
- Randerson-Moor, J.A.; Harland, M.; Williams, S.; Cuthbert-Heavens, D.; Sheridan, E.; Aveyard, J.; Sibley, K.; Whitaker, L.; Knowles, M.; Bishop, J.N.; et al. A germline deletion of p14ARF but not cdkn2a in a melanoma-neural system tumour syndrome family. Hum. Mol. Genet. 2001, 10, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sakaki, T.; Hashimoto, H.; Nakase, H.; Ishida, E.; Shimada, K.; Konishi, N. Frequent alterations of the p14arf and p16ink4a genes in primary central nervous system lymphomas. Cancer Res. 2001, 61, 6335–6339. [Google Scholar] [PubMed]
- Silva, J.; Dominguez, G.; Silva, J.M.; Garcia, J.M.; Gallego, I.; Corbacho, C.; Provencio, M.; Espana, P.; Bonilla, F. Analysis of genetic and epigenetic processes that influence p14ARF expression in breast cancer. Oncogene 2001, 20, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Vonlanthen, S.; Heighway, J.; Tschan, M.P.; Borner, M.M.; Altermatt, H.J.; Kappeler, A.; Tobler, A.; Fey, M.F.; Thatcher, N.; Yarbrough, W.G.; et al. Expression of p16ink4a/p16alpha and p19ARF/p16β is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene 1998, 17, 2779–2785. [Google Scholar] [CrossRef] [PubMed]
- Palmero, I.; Pantoja, C.; Serrano, M. P19arf links the tumour suppressor p53 to Ras. Nature 1998, 395, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, E.; Lamed, Y.; Ginsberg, D. E2F and Ras synergize in transcriptionally activating p14arf expression. Cell Cycle 2003, 2, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Parisi, T.; Pollice, A.; Di Cristofano, A.; Calabro, V.; La Mantia, G. Transcriptional regulation of the human tumor suppressor p14ARF by e2f1, e2f2, e2f3, and sp1-like factors. Biochem. Biophys. Res. Commun. 2002, 291, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. MDM2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase e3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by MDM2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Tsai, K.Y.; Jacks, T. An intact HDM2 ring-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2000, 2, 563–568. [Google Scholar] [CrossRef] [PubMed]
- MarikoOkada-Hatakeyama, S.M.K.I. Current status of mathematical modeling of cancer—From the viewpoint of cancer hallmarks. Curr. Opin. Syst. Biol. 2017, 2, 39–48. [Google Scholar]
- Chen, J.; Wu, X.; Lin, J.; Levine, A.J. MDM-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol. Cell. Biol. 1996, 16, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The INK4a tumor suppressor gene product, p19ARf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and MDM2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Levine, A.J. P19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of MDM2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell 1999, 3, 579–591. [Google Scholar] [CrossRef]
- Rizos, H.; Darmanian, A.P.; Mann, G.J.; Kefford, R.F. Two arginine rich domains in the p14ARF tumour suppressor mediate nucleolar localization. Oncogene 2000, 19, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Ashcroft, M.; Kubbutat, M.H.; Vousden, K.H. Contribution of two independent MDM2-binding domains in p14ARF to p53 stabilization. Curr. Biol. 2000, 10, 539–542. [Google Scholar] [CrossRef]
- Weber, J.D.; Kuo, M.L.; Bothner, B.; DiGiammarino, E.L.; Kriwacki, R.W.; Roussel, M.F.; Sherr, C.J. Cooperative signals governing ARF-MDM2 interaction and nucleolar localization of the complex. Mol. Cell. Biol. 2000, 20, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar ARF sequesters MDM2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19ARF with MDM2 inhibits ubiquitin ligase activity of MDM2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Midgley, C.A.; Desterro, J.M.; Saville, M.K.; Howard, S.; Sparks, A.; Hay, R.T.; Lane, D.P. An N-terminal p14ARF peptide blocks MDM2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 2000, 19, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Llanos, S.; Peters, G. Multiple interacting domains contribute to p14ARF mediated inhibition of MDM2. Oncogene 2002, 21, 4498–4507. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, C.; Zhao, L.; Modestou, M.; Quelle, D.E. ARF function does not require p53 stabilization or MDM2 relocalization. Mol. Cell. Biol. 2002, 22, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. P53-independent functions of the p19ARF tumor suppressor. Genes Dev. 2000, 14, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Leduc, C.; Coll, J.L.; Brambilla, E.; Gazzeri, S. P14arf induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 2003, 22, 1822–1835. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Vivo, M.; La Mantia, G. The promiscuity of ARF interactions with the proteasome. FEBS Lett. 2008, 582, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.; Bartkova, J.; Mistrik, M.; Hall, A.; Lange, M.K.; Ralfkiaer, U.; Bartek, J.; Guldberg, P. A short acidic motif in ARF guards against mitochondrial dysfunction and melanoma susceptibility. Nat. Commun. 2014, 5, 5348. [Google Scholar] [CrossRef] [PubMed]
- Britigan, E.M.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol. Biol. Cell 2014, 25, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, V.S.; Hagen, J.; Frazier, A.A.; Lushnikova, T.; Fitzgerald, M.P.; di Tommaso, A.; Ladeveze, V.; Domann, F.E.; Eischen, C.M.; Quelle, D.E. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J. Biol. Chem. 2007, 282, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; den Besten, W.; Thomas, M.C.; Sherr, C.J. ARF-induced turnover of the nucleolar nucleophosmin-associated sumo-2/3 protease senp3. Cell Cycle 2008, 7, 3378–3387. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the ARF tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Chisholm, J.; Desterro, J.M.; Lane, D.P.; Hay, R.T. P14ARF promotes accumulation of sumo-1 conjugated (h)MDM2. FEBS Lett. 2002, 528, 207–211. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La Mantia, G. P14arf interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed]
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the ARF tumor suppressor protein with nucleophosmin/b23. Mol. Cell. Biol. 2004, 24, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Bertwistle, D.; Den Besten, W.; Kuo, M.L.; Sugimoto, M.; Tago, K.; Williams, R.T.; Zindy, F.; Roussel, M.F. p53-dependent and -independent functions of the ARF tumor suppressor. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Dwertmann, A.; Herold, S.; Abed, M.; Naud, J.F.; Finkernagel, F.; Harms, G.S.; Orian, A.; Wanzel, M.; Eilers, M. The ARF tumor suppressor protein inhibits miz1 to suppress cell adhesion and induce apoptosis. J. Cell Biol. 2010, 188, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses sumo modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Santockyte, R.; Shen, R.F.; Tekle, E.; Wang, G.; Yang, D.C.; Chock, P.B. Expression of sumo-2/3 induced senescence through p53- and pRb-mediated pathways. J. Biol. Chem. 2006, 281, 36221–36227. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J. MDM2-ARF complex regulates p53 sumoylation. Oncogene 2003, 22, 5348–5357. [Google Scholar] [CrossRef] [PubMed]
- Damalas, A.; Velimezi, G.; Kalaitzakis, A.; Liontos, M.; Papavassiliou, A.G.; Gorgoulis, V.; Angelidis, C. Loss of p14ARF confers resistance to heat shock- and oxidative stress-mediated cell death by upregulating beta-catenin. Int. J. Cancer 2011, 128, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Fatyol, K.; Szalay, A.A. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1α (hif-1α) and inhibits hif-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Festa, L.; Duverger, O.; Vivo, M.; Guerrini, L.; La Mantia, G.; Morasso, M.I.; Calabro, V. Homeodomain protein dlx3 induces phosphorylation-dependent p63 degradation. Cell Cycle 2009, 8, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Festa, L.; Roscigno, G.; Vivo, M.; Pollice, A.; Morasso, M.; La Mantia, G.; Calabro, V. A dominant mutation etiologic for human tricho-dento-osseous syndrome impairs the ability of DLX3 to downregulate ΔNp63α. J. Cell. Physiol. 2011, 226, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Calabro, V.; Mansueto, G.; Santoro, R.; Gentilella, A.; Pollice, A.; Ghioni, P.; Guerrini, L.; La Mantia, G. Inhibition of p63 transcriptional activity by p14ÂRF: Functional and physical link between human ARF tumor suppressor and a member of the p53 family. Mol. Cell. Biol. 2004, 24, 8529–8540. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Di Costanzo, A.; Fortugno, P.; Pollice, A.; Calabro, V.; La Mantia, G. Downregulation of ΔNp63α in keratinocytes by p14ARF-mediated sumo-conjugation and degradation. Cell Cycle 2009, 8, 3545–3551. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Vivo, M.; De Simone, M.; Guerrini, L.; Pollice, A.; La Mantia, G.; Calabro, V. Sumoylation and ubiquitylation crosstalk in the control of Δnp63α protein stability. Gene 2018, 645, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Guarino, A.M.; Troiano, A.; Pizzo, E.; Bosso, A.; Vivo, M.; Pinto, G.; Amoresano, A.; Pollice, A.; La Mantia, G.; Calabro, V. Oxidative stress causes enhanced secretion of YB-1 protein that restrains proliferation of receiving cells. Genes 2018, 9, 513. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, O.; Troiano, A.; Guarino, A.M.; Pollice, A.; Vivo, M.; La Mantia, G.; Calabro, V. Δnp63α controls YB-1 protein stability: Evidence on YB-1 as a new player in keratinocyte differentiation. Genes Cells 2016, 21, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Troiano, A.; Lomoriello, I.S.; di Martino, O.; Fusco, S.; Pollice, A.; Vivo, M.; La Mantia, G.; Calabro, V. Y-box binding protein-1 is part of a complex molecular network linking Δnp63α to the PI3K/AKT pathway in cutaneous squamous cell carcinoma. J. Cell. Physiol. 2015, 230, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Apicelli, A.J.; Maggi, L.B., Jr.; Hirbe, A.C.; Miceli, A.P.; Olanich, M.E.; Schulte-Winkeler, C.L.; Saporita, A.J.; Kuchenreuther, M.; Sanchez, J.; Weilbaecher, K.; et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol. Cell. Biol. 2008, 28, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Saporita, A.J.; Maggi, L.B., Jr.; Apicelli, A.J.; Weber, J.D. Therapeutic targets in the ARF tumor suppressor pathway. Curr. Med. Chem. 2007, 14, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar ARF tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Ayrault, O.; Andrique, L.; Larsen, C.J.; Seite, P. Human ARF tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene 2004, 23, 8097–8104. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the ARF tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Rodway, H.; Llanos, S.; Rowe, J.; Peters, G. Stability of nucleolar versus non-nucleolar forms of human p14ARF. Oncogene 2004, 23, 6186–6192. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor suppressor ARF degrades b23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (b23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Bonetti, P.; Lazzerini Denchi, E.; Martinelli, P.; Zamponi, R.; Marine, J.C.; Helin, K.; Falini, B.; Pelicci, P.G. Nucleophosmin is required for DNA integrity and p19ARF protein stability. Mol. Cell. Biol. 2005, 25, 8874–8886. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Grisendi, S.; Clohessy, J.G.; Majid, S.; Bernardi, R.; Sportoletti, P.; Pandolfi, P.P. The leukemia-associated cytoplasmic nucleophosmin mutant is an oncogene with paradoxical functions: ARF inactivation and induction of cellular senescence. Oncogene 2007, 26, 7391–7400. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Chiarella, S.; Franceschini, M.; Di Matteo, A.; Brunori, M.; Banci, L.; Federici, L. Identification of a novel nucleophosmin-interaction motif in the tumor suppressor p14ARF. FEBS J. 2018, 285, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Martinelli, P.; Zamponi, R.; Shing, D.C.; Bonetti, P.; Luzi, L.; Volorio, S.; Bernard, L.; Pruneri, G.; Alcalay, M.; et al. Delocalization and destabilization of the ARF tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006, 66, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, W.; Kuo, M.L.; Tago, K.; Williams, R.T.; Sherr, C.J. Ubiquitination of, and sumoylation by, the ARF tumor suppressor. ISR Med. Assoc. J. 2006, 8, 249–251. [Google Scholar] [PubMed]
- Kuo, M.L.; den Besten, W.; Sherr, C.J. N-terminal polyubiquitination of the ARF tumor suppressor, a natural lysine-less protein. Cell Cycle 2004, 3, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Nasti, V.; Ronca, R.; Vivo, M.; Lo Iacono, M.; Calogero, R.; Calabro, V.; La Mantia, G. Functional and physical interaction of the human ARF tumor suppressor with TAT-binding protein-1. J. Biol. Chem. 2004, 279, 6345–6353. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K.; Bengal, E.; Ziv, T.; Admon, A.; Ciechanover, A. A novel site for ubiquitination: The N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998, 17, 5964–5973. [Google Scholar] [CrossRef] [PubMed]
- Sheaff, R.J.; Singer, J.D.; Swanger, J.; Smitherman, M.; Roberts, J.M.; Clurman, B.E. Proteasomal turnover of p21cip1 does not require p21cip1 ubiquitination. Mol. Cell 2000, 5, 403–410. [Google Scholar] [CrossRef]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The trip from ULF to ARF. Cancer Cell 2010, 17, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Sepe, M.; Villella, V.R.; Tolino, F.; Vivo, M.; Calabro, V.; La Mantia, G. Tbp-1 protects the human oncosuppressor p14ARF from proteasomal degradation. Oncogene 2007, 26, 5154–5162. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGγ proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the INK4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16INK4a and p19ARF senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. INK4-ARF locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Williams, R.T.; Baudino, T.A.; Rehg, J.E.; Skapek, S.X.; Cleveland, J.L.; Roussel, M.F.; Sherr, C.J. ARF tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 15930–15935. [Google Scholar] [CrossRef] [PubMed]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.L.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of PDGF signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the ARF tumor suppressor during male germ cell and eye development in ARF-CRE reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Mei, J.; Liu, J.; Skapek, S.X. Mir-34a is essential for p19ARF-driven cell cycle arrest. Cell Cycle 2014, 13, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Finkelstein, D.; Sherr, C.J. ARF tumor suppressor and Mir-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1112–E1121. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Roig, I.; Jasin, M.; Keeney, S.; Sherr, C.J. Expression of ARF tumor suppressor in spermatogonia facilitates meiotic progression in male germ cells. PLoS Genet. 2011, 7, e1002157. [Google Scholar] [CrossRef] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Kotsinas, A.; Papanagnou, P.; Evangelou, K.; Trigas, G.C.; Kostourou, V.; Townsend, P.; Gorgoulis, V.G. ARF: A versatile DNA damage response ally at the crossroads of development and tumorigenesis. Front. Genet. 2014, 5, 236. [Google Scholar] [CrossRef] [PubMed]
- Bieging-Rolett, K.T.; Johnson, T.M.; Brady, C.A.; Beaudry, V.G.; Pathak, N.; Han, S.; Attardi, L.D. p19ARF is required for the cellular response to chronic DNA damage. Oncogene 2016, 35, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Deyrieux, A.F.; Rosas-Acosta, G.; Ozbun, M.A.; Wilson, V.G. Sumoylation dynamics during keratinocyte differentiation. J. Cell Sci. 2007, 120, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Cho, M.S.; Gi, Y.J.; Ayanga, B.A.; Sherr, C.J.; Flores, E.R. Rescue of key features of the p63-null epithelial phenotype by inactivation of INK4a and ARF. EMBO J. 2009, 28, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aguilera, A.; Sanchez-Beato, M.; Garcia, J.F.; Prieto, I.; Pollan, M.; Piris, M.A. p14ARF nuclear overexpression in aggressive B-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Park, J.Y.; Kang, H.J.; Cho, H.C. Overexpression of p16INK4a and p14ARF in haematological malignancies. Clin. Lab. Haematol. 2003, 25, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of CDKN2a α (p16INK4a) and β (p14ARF) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Humbey, O.; Pimkina, J.; Zilfou, J.T.; Jarnik, M.; Dominguez-Brauer, C.; Burgess, D.J.; Eischen, C.M.; Murphy, M.E. The ARF tumor suppressor can promote the progression of some tumors. Cancer Res. 2008, 68, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Vivo, M. Dynamics of p14ARF and focal adhesion kinase-mediated autophagy in cancer. Cancers 2018, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14ARF tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kimmelman, A.C. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, M.; Burigo, L.; Durante, M.; Herrlitz, M.; Kramer, M.; Mishustin, I.; Muller, I.; Natale, F.; Pshenichnov, I.; Schramm, S.; et al. Nanolesions induced by heavy ions in human tissues: Experimental and theoretical studies. Beilstein J. Nanotechnol. 2012, 3, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Dallaglio, K.; Torquato, P.; Piroddi, M.; Galli, F. NRF2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018, 193, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Horl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Scholl, A.; Rapp, A.; Yu, W.; Rausch, C.; Cardoso, M.C. DNA replication and repair kinetics of ALU, LINE1 and satellite III genomic repetitive elements. Epigenet. Chromatin 2018, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Gu, W. p53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Carracedo, A.; Lin, H.K.; Koutcher, J.A.; Behrendt, N.; Egia, A.; Alimonti, A.; Carver, B.S.; Gerald, W.; Teruya-Feldstein, J.; et al. Differential p53-independent outcomes of p19ARF loss in oncogenesis. Sci. Signal. 2009, 2, ra44. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19ARF in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Federico Bocci, S.C.T.; Mercedes, S.V.; George, J.T.; Casabar, J.; Wong, P.K.; Hanash, S.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Nrf2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. bioRxiv 2018. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. MMP7 interacts with ARF in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Wang, S.C.; Morris, J.P.T.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.C.; Cheung, S.T.; Fan, S.T. Atypical localization of membrane type 1-matrix metalloproteinase in the nucleus is associated with aggressive features of hepatocellular carcinoma. Mol. Carcinog. 2007, 46, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Berggren, P.; Kumar, R.; Sakano, S.; Hemminki, L.; Wada, T.; Steineck, G.; Adolfsson, J.; Larsson, P.; Norming, U.; Wijkstrom, H.; et al. Detecting homozygous deletions in the cdkn2a(p16(ink4a))/arf(p14(arf)) gene in urinary bladder cancer using real-time quantitative PCR. Clin. Cancer Res. 2003, 9, 235–242. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, J.P.; Cha, E.K.; Iyer, G.; Scott, S.N.; Zabor, E.C.; Shah, R.H.; Ren, Q.; Bagrodia, A.; Kim, P.H.; Hakimi, A.A.; et al. Genomic characterization of upper tract urothelial carcinoma. Eur. Urol. 2015, 68, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, T.B.; Kobayashi, T.; Ramirez, R.; Rong, L.; Puzio-Kuter, A.M.; Iyer, G.; Teo, M.Y.; Sanchez-Vega, F.; Wang, J.; Schultz, N.; et al. ARF confers a context-dependent response to chemotherapy in muscle-invasive bladder cancer. Cancer Res. 2017, 77, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.C.; Wang, W.J.; Yao, C.C.; Wu, P.R.; Chen, R.H. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J. Cell Biol. 2006, 172, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Kimchi, A. A smarf way to die: A novel short isoform of p19ARF is linked to autophagic cell death. Autophagy 2006, 2, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; Kontogiannea, M.; Fon, E.A. Short mitochondrial ARF triggers parkin/pink1-dependent mitophagy. J. Biol. Chem. 2014, 289, 29519–29530. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase JNK2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor arf for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.S.B.; McEwan, D.G.; Morton, J.P.; Macagno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; Sansom, O.J.; et al. Autophagic targeting of SRC promotes cancer cell survival following reduced fak signalling. Nat. Cell Biol. 2011, 14, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kadandale, P.; Kiger, A.A. Role of selective autophagy in cellular remodeling: “Self-eating” into shape. Autophagy 2010, 6, 1194–1195. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Novel insights into how autophagy regulates tumor cell motility. Autophagy 2016, 12, 1679–1680. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Ranieri, M.; Sansone, F.; Santoriello, C.; Calogero, R.A.; Calabro, V.; Pollice, A.; La Mantia, G. Mimicking p14ARF phosphorylation influences its ability to restrain cell proliferation. PLoS ONE 2013, 8, e53631. [Google Scholar] [CrossRef]
- Vivo, M.; Matarese, M.; Sepe, M.; Di Martino, R.; Festa, L.; Calabro, V.; La Mantia, G.; Pollice, A. Mdm2-mediated degradation of p14arf: A novel mechanism to control ARF levels in cancer cells. PLoS ONE 2015, 10, e0117252. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Guidone, D.; Sangermano, F.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. PKC dependent p14ARF phosphorylation on threonine 8 drives cell proliferation. Sci. Rep. 2018, 8, 7056. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Functions | Mechanisms |
|---|---|
| Tumor Suppression | |
| Growth control | p53 dependent and independent call cycle arrest and apoptosis. ΔNp63 inhibition [4,5,11,12,13,31,32,33,34,42,43,55,79,80] |
| Chromosomal stability | Stabilization of miotic spindle, prevents aneuploidy [62] |
| Ribosomal biogenesis | Inhibition or rRNA processing and transcription [84,85,86,87] |
| Oxidative stress | Cellular protection from dysfunctional mitochondria [61] |
| DNA damage | Activation of ATM/ATR/CHK pathway, p53-dependent pathways of DNA repair [115,116] |
| Contest Dependent | |
| Autophagy | Beclin-1 activation, dissipation of mitochondrion potential [60,123,124,125,126,127] |
| Differentiation | Inhibits angiogenesis in developing eye, protects from apoptosis in spermatogonia, allows extraembryonic endoderm migration [79,110,111,112,113,114,115,116,117] |
| Tumor Promoter | |
| Epithelial to mesechimal transition | Slug stabilization [139] |
| Modulation of tumor microenvironment | Metallopeptidase-1 interaction [141] |
| Survival of lymphoma, protease and bladder tumor cells | Promotes prostate cancer in Pten mouse model and autophagy in lymphomas; promotes chemoresistance in bladder cancer [120,121,122,123,124,125,126,127,138,141,147] |
| Anoikis protection, increased proliferation | Inhibition of DAPK mediated cell death; activation focal adhesion kinase [67,160,161,162] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. https://doi.org/10.3390/biom9030087
Fontana R, Ranieri M, La Mantia G, Vivo M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules. 2019; 9(3):87. https://doi.org/10.3390/biom9030087
Chicago/Turabian StyleFontana, Rosa, Michela Ranieri, Girolama La Mantia, and Maria Vivo. 2019. "Dual Role of the Alternative Reading Frame ARF Protein in Cancer" Biomolecules 9, no. 3: 87. https://doi.org/10.3390/biom9030087
APA StyleFontana, R., Ranieri, M., La Mantia, G., & Vivo, M. (2019). Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules, 9(3), 87. https://doi.org/10.3390/biom9030087