Attenuation of Glucose-Induced Myoglobin Glycation and the Formation of Advanced Glycation End Products (AGEs) by (R)-α-Lipoic Acid In Vitro


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Evaluation of the Inhibitory Effect of α-Lipoic Acid on Myoglobin Glycation under Glucose Overload In Vitro
2.2.1. Determination of Fluorescent AGEs Formation
2.2.2. Estimation of Free Iron in Glycated Myoglobin (Ferrozine Test)
2.2.3. Estimation of Fructosamine (Glycated Myoglobin)
2.2.4. Estimation of Protein Carbonyls Content
2.2.5. Estimation of Free Protein Thiols
2.2.6. Statistical Analysis
3. Results
3.1. Effect of α-Lipoic Acid on the Formation of Fluorescent AGEs
3.2. Effect of α-Lipoic Acid on Free Iron Release
3.3. Effect of α-Lipoic Acid on Fructosamine Formation
3.4. Effect of α-Lipoic Acid on Protein Carbonyls Formation
3.5. Effect of α-Lipoic Acid on Protein Thiols Oxidation
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ordway, G.A.; Garry, D.J. Myoglobin: An essential hemoprotein in striated muscle. J. Exp. Biol. 2004, 207, 3441–3446. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Huang, S.-J.; Glabe, A.; Jue, T. Implication of CO inactivation on myoglobin function. Am. J. Physiol. Cell Physiol. 2006, 290, C1616–C1624. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, K.D.; Papadopoulos, S.; Peters, T.; Gros, G. Myoglobin: Just an oxygen store or also an oxygen transporter. News Physiol. Sci. 2000, 15, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Kreutzer, U.; Jue, T. Myoglobin translational diffusion in rat myocardium and its implication on intracellular oxygen transport. J. Physiol. 2007, 578, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Merx, M.W.; Flögel, U.; Stumpe, T.; Gödecke, A.; Decking, U.K.; Schrader, J. Myoglobin facilitates oxygen diffusion. FASEB J. 2001, 15, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Flogel, U.; Merx, M.W.; Godecke, A.; Decking, U.K.; Schrader, J. Myoglobin: A scavenger of bioactive NO. Proc. Natl. Acad. Sci. USA 2001, 98, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Brunori, M. Nitric oxide moves myoglobin centre stage. Trends Biochem. Sci. 2001, 26, 209–210. [Google Scholar] [CrossRef]
- Wunderlich, C.; Flogel, U.; Godecke, A.; Heger, J.; Schrader, J. Acute inhibition of myoglobin impairs contractility and energy state of iNOS-overexpressing hearts. Circ. Res. 2003, 92, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Sen, S.; Chakraborti, A.S. In vitro nonenzymatic glycation enhances the role of myoglobin as a source of oxidative stress. Free Radic. Res. 2004, 38, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Hendgen-Cotta, U.B.; Kelm, M.; Rassaf, T. Myoglobin functions in the heart. Free Radic. Biol. Med. 2014, 73, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ames, J.M.; Smith, R.D.; Baynes, J.W.; Metz, T.O. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: Probing the pathogenesis of chronic disease. J. Proteome Res. 2009, 8, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Chakraborti, A.S. In vitro study on structural alteration of myoglobin by methylglyoxal. Protein J. 2013, 32, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Sil, R.; Chakraborti, A.S. Non-enzymatic glycation induces structural modifications of myoglobin. Mol. Cell. Biochem. 2010, 338, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rosen, P.; Nawroth, P.P.; King, G.; Moller, W.; Tritschler, H.J.; Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: A summary of a congress series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, K.W.; Kaiser, H.; Grone, H.J.; Trapp, V.E.; Meinck, H.M.; Goebel, H.H.; Kunze, E.; Kreuzer, H.; Scheler, F. Myoglobinuric renal failure in hyperosmolar diabetic coma. Dtsch. Med. Wochenschr. 1981, 106, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Mugikura, M.; Endoh, M.; Ogami, Y.; Otsuki, M. Acute pancreatitis with diabetic ketoacidosis associated with hypermyoglobinemia, acute renal failure and DIC. J. Gastroenterol. 1996, 31, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Nguyen, N.; Schwaiger, M. Myocardial glucose transporter GLUT1: Translocation induced by insulin and ischemia. J. Mol. Cell. Cardiol. 1999, 31, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Tian, R. Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [PubMed]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G.; Baynes, J.W.; Hudson, B.G. Amadorins: Novel post-Amadori inhibitors of advanced glycation reactions. Biochem. Biophys. Res. Commun. 1999, 257, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.O. Biological effects of aminoguanidine: An update. Inflamm. Res. 1999, 48, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 1988, 318, 1315–1321. [Google Scholar] [PubMed]
- Brownlee, M. Lilly lecture 1993. Glycation and diabetic complications. Diabetes 1994, 43, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Vasan, S.; Foiles, P.; Founds, H. Therapeutic potential of breakers of advanced glycation end product-protein crosslinks. Arch. Biochem. Biophys. 2003, 419, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Witt, E.H.; Tritschler, H.J. α-lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. α-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Goraca, A.; Huk-Kolega, H.; Piechota, A.; Kleniewska, P.; Ciejka, E.; Skibska, B. Lipoic acid—Biological activity and therapeutic potential. Pharmacol. Rep. 2011, 63, 849–858. [Google Scholar] [CrossRef]
- Ghelani, H.; Razmovski-Naumovski, V.; Nammi, S. Chronic treatment of (R)-α-lipoic acid reduces blood glucose and lipid levels in high-fat diet and low-dose streptozotocin-induced metabolic syndrome and type 2 diabetes in Sprague-Dawley rats. Pharmacol. Res. Perspect. 2017, 5, e00306. [Google Scholar] [CrossRef] [PubMed]
- Midaoui, A.E.; Elimadi, A.; Wu, L.; Haddad, P.S.; de Champlain, J. Lipoic acid prevents hypertension, hyperglycemia and the increase in heart mitochondrial superoxide production. Am. J. Hypertens 2003, 16, 173–179. [Google Scholar] [CrossRef]
- Thirunavukkarasu, V.; Nandhini, A.T.; Anuradha, C.V. Fructose diet-induced skin collagen abnormalities are prevented by lipoic acid. Exp. Diabesity Res. 2004, 5, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, V.; Anitha Nandhini, A.T.; Anuradha, C.V. Lipoic acid improves glucose utilisation and prevents protein glycation and age formation. Pharmazie 2005, 60, 772–775. [Google Scholar] [PubMed]
- Muellenbach, E.M.; Diehl, C.J.; Teachey, M.K.; Lindborg, K.A.; Hasselwander, O.; Matuschek, M.; Henriksen, E.J. Metabolic interactions of AGE inhibitor pyridoxamine and antioxidant α-lipoic acid following 22 weeks of treatment in obese Zucker rats. Life Sci. 2009, 84, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Yan, H.D.; Wang, J. Extract of Ginkgo biloba and α-lipoic acid attenuate advanced glycation end products accumulation and rage expression in diabetic nephropathy rats. Zhongguo Zhong Xi Yi Jie He Za Zhi 2011, 31, 525–531. (In Chinese) [Google Scholar] [PubMed]
- Leu, J.G.; Lin, C.Y.; Jian, J.H.; Shih, C.Y.; Liang, Y.J. Epigallocatechin-3-gallate combined with α lipoic acid attenuates high glucose-induced receptor for advanced glycation end products (RAGE) expression in human embryonic kidney cells. An. Acad. Bras. Ciênc. 2013, 85, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 2007, 148, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Chen, H.M.; Yao, Y.D.; Hung, C.F.; Tu, C.S.; Liang, Y.J. Topical treatment with anti-oxidants and Au nanoparticles promote healing of diabetic wound through receptor for advance glycation end-products. Eur. J. Pharm. Sci. 2012, 47, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Akihiko, S.M.Y.; Satoshi, H.; Takayuki, N.; Masafumi, I.; Seigo, K.; Yoshikazu, Y. Anti-glycation activity of α-lipoic acid derivatives and vitamin E derivatives. Anti-Aging Med. 2013, 10, 42–54. [Google Scholar]
- Suzuki, Y.J.; Tsuchiya, M.; Packer, L. Lipoate prevents glucose-induced protein modifications. Free Radic. Res. Commun. 1992, 17, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Chevion, S.; Chevion, M.; Hofmann, M.; Quehenberger, P.; Illmer, T.; Luther, T.; Berentshtein, E.; Tritschler, H.; Muller, M.; et al. Advanced glycation end product-induced activation of NF-κB is suppressed by α-lipoic acid in cultured endothelial cells. Diabetes 1997, 46, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Effect of advanced glycation end products on accelerated apoptosis of retinal capillary cells under in vitro conditions. Life Sci. 2005, 76, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Kunt, T.; Forst, T.; Wilhelm, A.; Tritschler, H.; Pfuetzner, A.; Harzer, O.; Engelbach, M.; Zschaebitz, A.; Stofft, E.; Beyer, J. α-lipoic acid reduces expression of vascular cell adhesion molecule-1 and endothelial adhesion of human monocytes after stimulation with advanced glycation end products. Clin. Sci. 1999, 96, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Schinzel, R.; Wiesinger, H.; Riederer, P.; Munch, G. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur. J. Neurosci. 2001, 14, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Gasic-Milenkovic, J.; Loske, C.; Munch, G. Advanced glycation endproducts cause lipid peroxidation in the human neuronal cell line SH-SY5Y. J. Alzheimers Dis. 2003, 5, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.Q.; Dong, C.F.; Dong, S.Q.; Dong, X.L.; Hong, Y.; Hou, X.Y.; Luo, D.Z.; Pei, J.J.; Liu, X.P. Ages induce cell death via oxidative and endoplasmic reticulum stresses in both human SH-Sy5Y neuroblastoma cells and rat cortical neurons. Cell. Mol. Neurobiol. 2012, 32, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Ghelani, H.; Razmovski-Naumovski, V.; Pragada, R.R.; Nammi, S. (R)-α-lipoic acid inhibits fructose-induced myoglobin fructation and the formation of advanced glycation end products (AGEs) in vitro. BMC Complement. Altern. Med. 2018, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, K.; Wrobel, K.; Garay-Sevilla, M.E.; Nava, L.E.; Malacara, J.M. Novel analytical approach to monitoring advanced glycosylation end products in human serum with on-line spectrophotometric and spectrofluorometric detection in a flow system. Clin. Chem. 1997, 43, 1563–1569. [Google Scholar] [PubMed]
- Panter, S.S. Release of iron from hemoglobin. Methods Enzymol. 1994, 231, 502–514. [Google Scholar] [PubMed]
- Ohkawara, E.; Nohara, Y.; Kanno, Y.; Suzuki, H.; Matsumoto, G.; Kinoshita, T.; Watanabe, M. Fructosamine assay using albumin extracted from serum. Biol. Pharm. Bull. 2002, 25, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Williams, J.A.; Stadtman, E.R.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994, 233, 346–357. [Google Scholar] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Wu, J.-W.; Hsieh, C.-L.; Wang, H.-Y.; Chen, H.-Y. Inhibitory effects of guava (Psidium guajava L.) leaf extracts and its active compounds on the glycation process of protein. Food Chem. 2009, 113, 78–84. [Google Scholar] [CrossRef]
- Elosta, A.; Ghous, T.; Ahmed, N. Natural products as anti-glycation agents: Possible therapeutic potential for diabetic complications. Curr. Diabetes Rev. 2012, 8, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Kanatous, S.B.; Mammen, P.P.; Rosenberg, P.B.; Martin, C.M.; White, M.D.; Dimaio, J.M.; Huang, G.; Muallem, S.; Garry, D.J. Hypoxia reprograms calcium signalling and regulates myoglobin expression. Am. J. Physiol. Cell Physiol. 2009, 296, C393–C402. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Gries, F.A. α-lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes 1997, 46, S62–S66. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Kang, C.H.; Wang, S.G.; Lee, H.M. α-lipoic acid regulates lipid metabolism through induction of sirtuin 1 (SIRT1) and activation of AMP-activated protein kinase. Diabetologia 2012, 55, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-Q.; Chen, Y.-Y.; Wang, X.; Li, X.-J.; Xue, B.; Qu, L.; Zhang, T.-T.; Mu, Y.-M.; Lu, J.-M. The protective effect of α lipoic acid on Schwann cells exposed to constant or intermittent high glucose. Biochem. Pharmacol. 2012, 84, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Muellenbach, E.A.; Diehl, C.J.; Teachey, M.K.; Lindborg, K.A.; Archuleta, T.L.; Harrell, N.B.; Andersen, G.; Somoza, V.; Hasselwander, O.; Matuschek, M.; et al. Interactions of the advanced glycation end product inhibitor pyridoxamine and the antioxidant α-lipoic acid on insulin resistance in the obese Zucker rat. Metabolism 2008, 57, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Kerkeni, M.; Saïdi, A.; Bouzidi, H.; Letaief, A.; Ben Yahia, S.; Hammami, M. Pentosidine as a biomarker for microvascular complications in type 2 diabetic patients. Diabetes Vasc. Dis. Res. 2013, 10, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, S.; Miyata, T.; Ueda, Y.; Tanaka, H.; Maeda, K.; Kawashima, S.; Van Ypersele de Strihou, C.; Kurokawa, K. Plasma levels of pentosidine in diabetic patients: An advanced glycation end product. J. Am. Soc. Nephrol. 1998, 9, 1681–1688. [Google Scholar] [PubMed]
- Weiss, M.F.; Rodby, R.A.; Justice, A.C.; Hricik, D.E. Free pentosidine and neopterin as markers of progression rate in diabetic nephropathy. Collaborative study group. Kidney Int. 1998, 54, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Okumura, K.; Aso, Y. High serum pentosidine concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism 2005, 54, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signalling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Bose, T.; Roy, D.; Chakraborti, A.S. Protoporphyrin IX-induced structural and functional changes in human red blood cells, haemoglobin and myoglobin. J. Biosci. 2004, 29, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Wollin, S.D.; Jones, P.J. α-lipoic acid and cardiovascular disease. J. Nutr. 2003, 133, 3327–3330. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Nakayama, Y.; Matsugo, S.; Kato, S. Protective effect of lipoic acid against oxidative stress is mediated by Keap1/Nrf2-dependent heme oxygenase-1 induction in the RGC-5 cellline. Brain Res. 2013, 1499, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Kurokawa, K.; Van Ypersele De Strihou, C. Advanced glycation and lipoxidation end products: Role of reactive carbonyl compounds generated during carbohydrate and lipid metabolism. J. Am. Soc. Nephrol. 2000, 11, 1744–1752. [Google Scholar] [PubMed]
- Zeng, J.; Dunlop, R.A.; Rodgers, K.J.; Davies, M.J. Evidence for inactivation of cysteine proteases by reactive carbonyls via glycation of active site thiols. Biochem. J. 2006, 398, 197–206. [Google Scholar] [CrossRef] [PubMed]
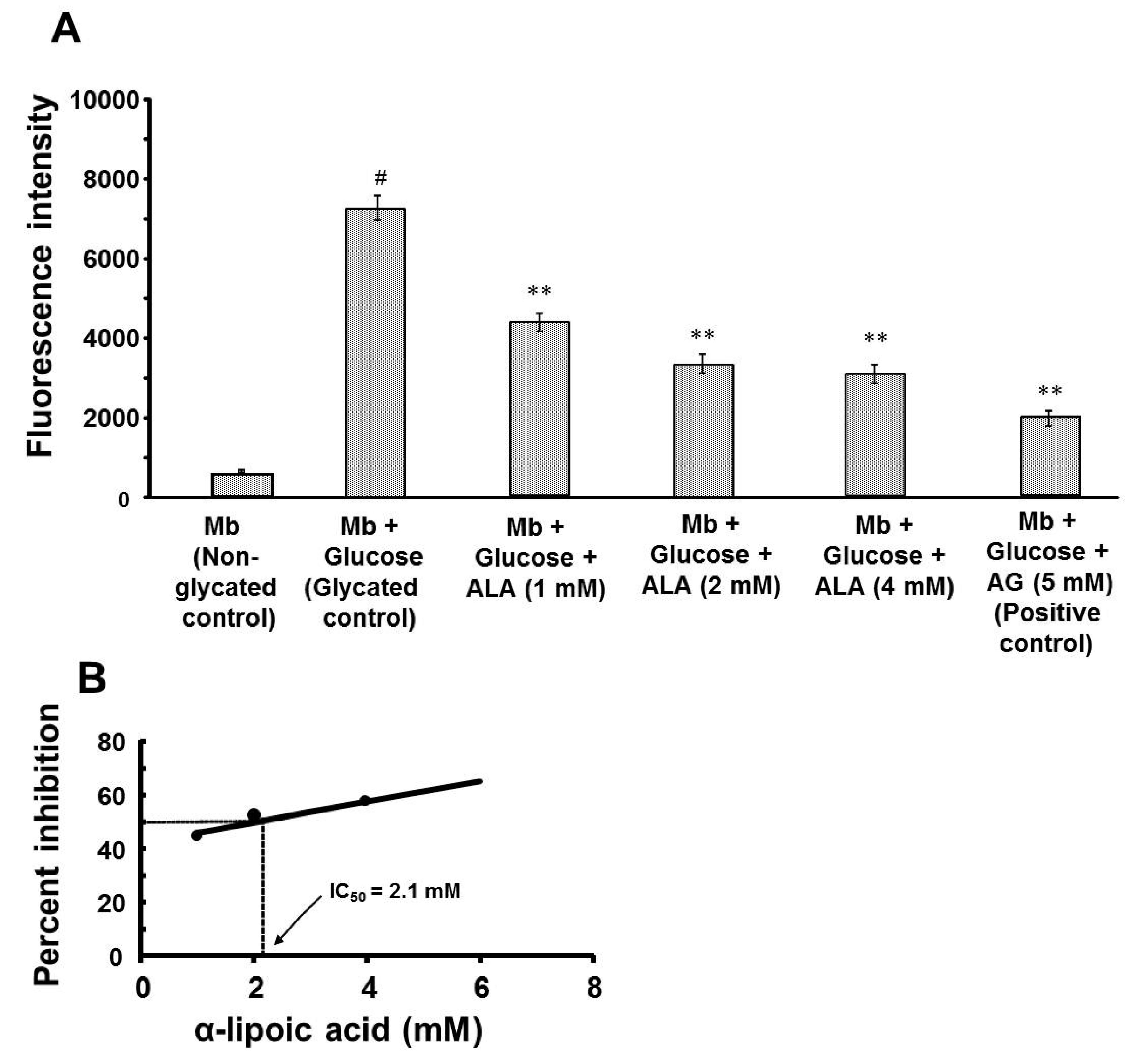

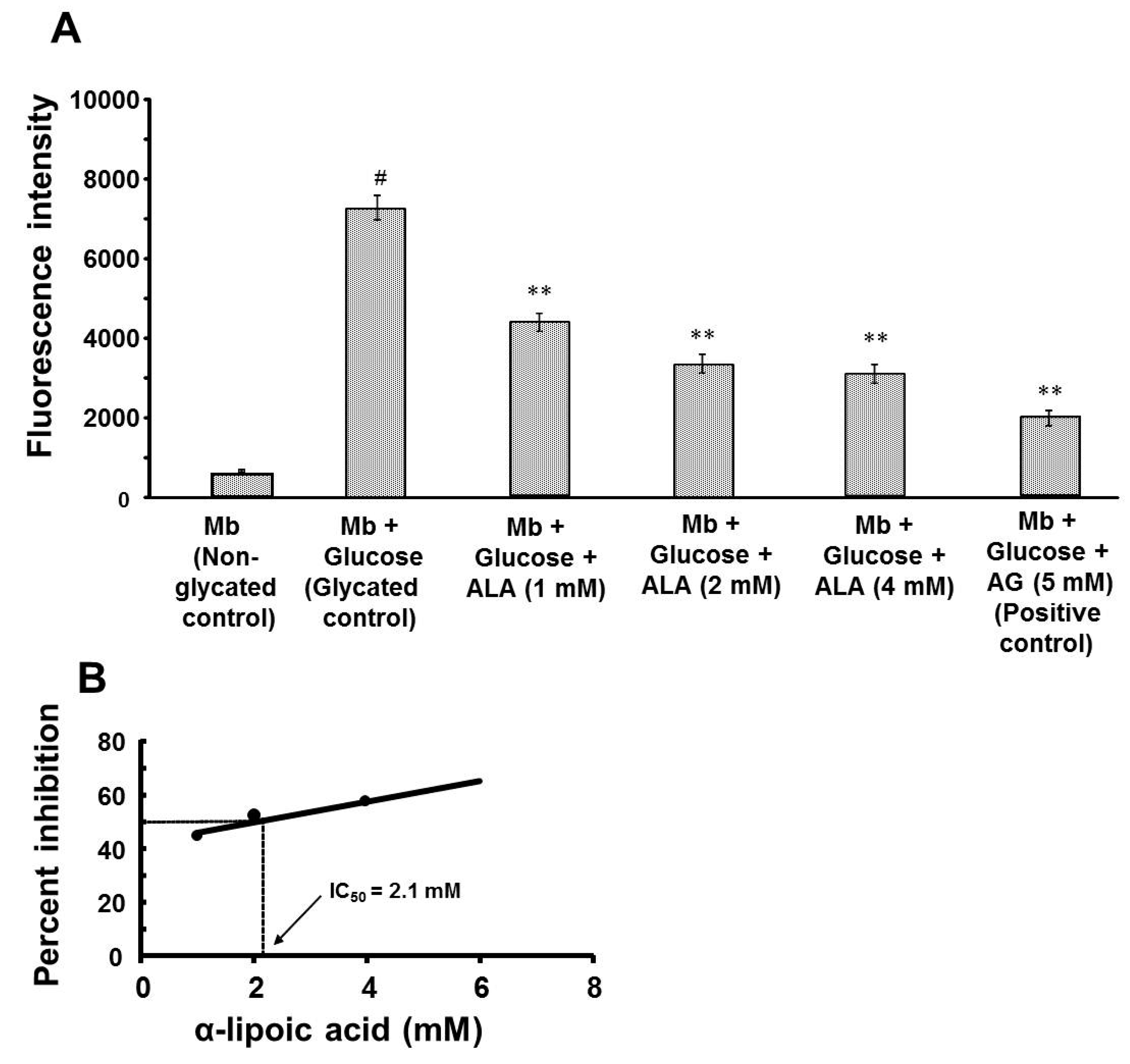{kind=link}
| Experimental Group | Concentration of Free Iron (µg/dL) | Percent Inhibition of Free Iron Release | IC50 (Day-30) | ||
|---|---|---|---|---|---|
| Day-10 | Day-20 | Day-30 | |||
| Mb (non-glycated control) | 9.4 ± 1.4 | 10.5 ± 1.3 | 11.5 ± 1.3 | - | 4.3 mM |
| Mb + Glu (glycated control) | 117.3 ± 0.4 ### | 122.0 ± 0.4 ### | 122.0 ± 0.5 ### | - | |
| Mb + Glu + ALA (1 mM) | 92.4 ± 0.3 * | 95.2 ± 0.3 ** | 95.3 ± 0.3 ** | 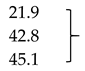 | |
| Mb + Glu + ALA (2 mM) | 68.4 ± 0.4 ** | 70.3 ± 0.6 ** | 69.8 ± 0.4 ** | ||
| Mb + Glu + ALA (4 mM) | 65.9 ± 0.5 ** | 66.7 ± 0.9 ** | 67.1 ± 0.2 ** | ||
| Mb + Glu + AG (5 mM; positive control) | 59.1 ± 0.5 ** | 61.0 ± 0.1 ** | 63.2 ± 2.0 ** | 48.2 | |
| Experimental Group | Fructosamine (µmol/L) | Percent Inhibition of Fructosamine Formation | IC50 (Day-30) | ||
|---|---|---|---|---|---|
| Day-10 | Day-20 | Day-30 | |||
| Mb (non-glycated control) | 486.4 ± 22.7 | 557.4 ± 13.5 | 564.9 ± 30.3 | - | 6.2 mM |
| Mb + Glu (glycated control) | 759.5 ± 21.6 # | 1131.8 ± 34.6 §,## | 1376.9 ± 27.8 §§,### | - | |
| Mb + Glu + ALA (1 mM) | 742.7 ± 18.2 | 959.7 ± 40.1 * | 1107.5 ± 13.1 ** | 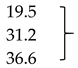 | |
| Mb + Glu + ALA (2 mM) | 617.3 ± 16.9 * | 795.0 ± 36.1 ** | 984.0 ± 11.3 ** | ||
| Mb + Glu + ALA (4 mM) | 501.3 ± 28.3 ** | 632.3 ± 71.1 ** | 871.8 ± 19.8 ** | ||
| Mb + Glu + AG (5 mM; positive control) | 445.2 ± 13.2 ** | 493.9 ± 21.4 ** | 606.1 ± 24.3 ** | 55.9 | |
| Experimental Group | Protein Carbonyls Content (nmol/mg Protein) | Percent Inhibition of Protein Carbonyls Formation | IC50 (Day-30) | ||
|---|---|---|---|---|---|
| Day-10 | Day-20 | Day-30 | |||
| Mb (non-glycated control) | 2.0 ± 0.4 | 2.3 ± 0.2 | 2.3 ± 0.2 | - | 3.4 mM |
| Mb + Glu (glycated control) | 5.4 ± 0.1 ### | 6.8 ± 0.3 ### | 8.2 ± 0.2 §,### | - | |
| Mb + Glu + ALA (1 mM) | 4.5 ± 0.1 * | 5.0. ± 0.1 * | 5.7 ± 0.1 ** | 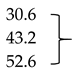 | |
| Mb + Glu + ALA (2 mM) | 3.9 ± 0.1 ** | 4.1 ± 0.2 ** | 4.4 ± 0.2 ** | ||
| Mb + Glu + ALA (4 mM) | 3.0 ± 0.1 ** | 3.2 ± 0.3 ** | 4.6 ± 0.1 ** | ||
| Mb + Glu + AG (5 mM; positive control) | 2.9 ± 0.1 ** | 3.0 ± 0.1 ** | 3.1 ± 0.2 ** | 60.6 | |
| Experimental Group | Free Protein Thiols (nmol/mg Protein) | Percent Increase of Protein Thiols | EC50 (Day-30) | ||
|---|---|---|---|---|---|
| Day-10 | Day-20 | Day-30 | |||
| Mb (non-glycated control) | 7.2 ± 0.4 | 6.9 ± 0.6 | 6.7 ± 0.3 | - | 3.4 mM |
| Mb + Glu (glycated control) | 2.8 ± 0.2 ### | 2.2 ± 0.1 ### | 1.9 ± 0.2 §,### | - | |
| Mb + Glu + ALA (1 mM) | 3.1 ± 0.7 | 2.7 ± 0.1 * | 2.5 ± 0.1 ** | 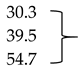 | |
| Mb + Glu + ALA (2 mM) | 3.2 ± 0.4 | 2.8 ± 0.6 * | 2.7 ± 0.5 ** | ||
| Mb + Glu + ALA (4 mM) | 3.5 ± 0.5 | 3.1 ± 0.3 ** | 3.0 ± 0.2 ** | ||
| Mb + Glu + AG (5 mM; positive control) | 4.6 ± 0.4 ** | 3.7 ± 0.1 ** | 3.4 ± 0.5 ** | 76.1 | |
| Anti-Glycation Assays | IC50/EC50 of ALA (mM) |
|---|---|
| Inhibition of fluorescent AGEs | 2.1 |
| Inhibition of fructosamine assay | 6.2 |
| Inhibition of free iron release | 4.3 |
| Inhibition of protein carbonyls | 3.4 |
| Protection of free protein thiols | 3.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghelani, H.; Razmovski-Naumovski, V.; Pragada, R.R.; Nammi, S. Attenuation of Glucose-Induced Myoglobin Glycation and the Formation of Advanced Glycation End Products (AGEs) by (R)-α-Lipoic Acid In Vitro. Biomolecules 2018, 8, 9. https://doi.org/10.3390/biom8010009
Ghelani H, Razmovski-Naumovski V, Pragada RR, Nammi S. Attenuation of Glucose-Induced Myoglobin Glycation and the Formation of Advanced Glycation End Products (AGEs) by (R)-α-Lipoic Acid In Vitro. Biomolecules. 2018; 8(1):9. https://doi.org/10.3390/biom8010009
Chicago/Turabian StyleGhelani, Hardik, Valentina Razmovski-Naumovski, Rajeswara Rao Pragada, and Srinivas Nammi. 2018. "Attenuation of Glucose-Induced Myoglobin Glycation and the Formation of Advanced Glycation End Products (AGEs) by (R)-α-Lipoic Acid In Vitro" Biomolecules 8, no. 1: 9. https://doi.org/10.3390/biom8010009
APA StyleGhelani, H., Razmovski-Naumovski, V., Pragada, R. R., & Nammi, S. (2018). Attenuation of Glucose-Induced Myoglobin Glycation and the Formation of Advanced Glycation End Products (AGEs) by (R)-α-Lipoic Acid In Vitro. Biomolecules, 8(1), 9. https://doi.org/10.3390/biom8010009