
Coupling TOR to the Cell Cycle by the Greatwall–Endosulfine–PP2A-B55 Pathway

Abstract
1. Introduction
2. Regulation of the G2/M Transition and Cell Size in Fission Yeast
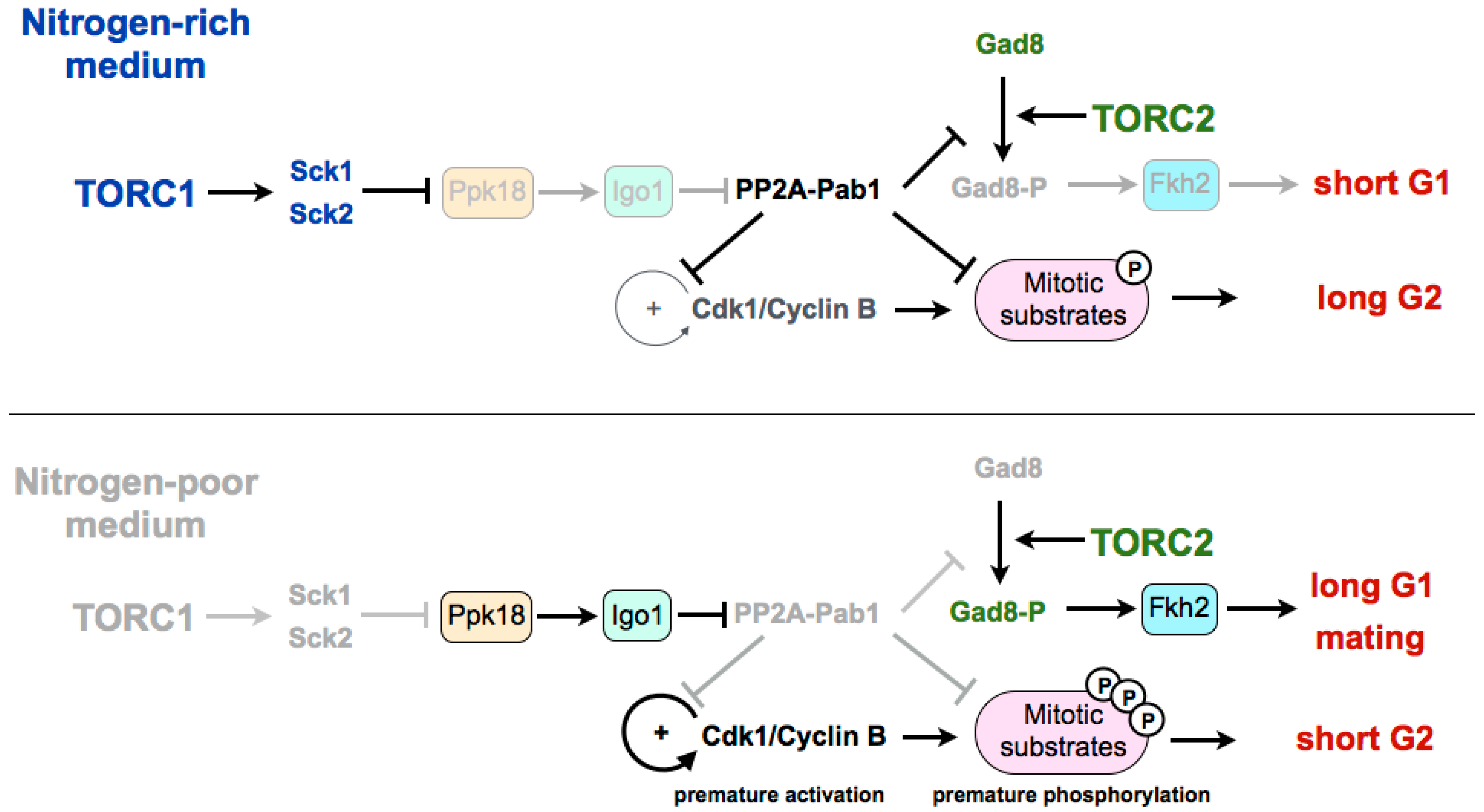
3. Coordination of TORC1 and TORC2 in the Differentiation Response
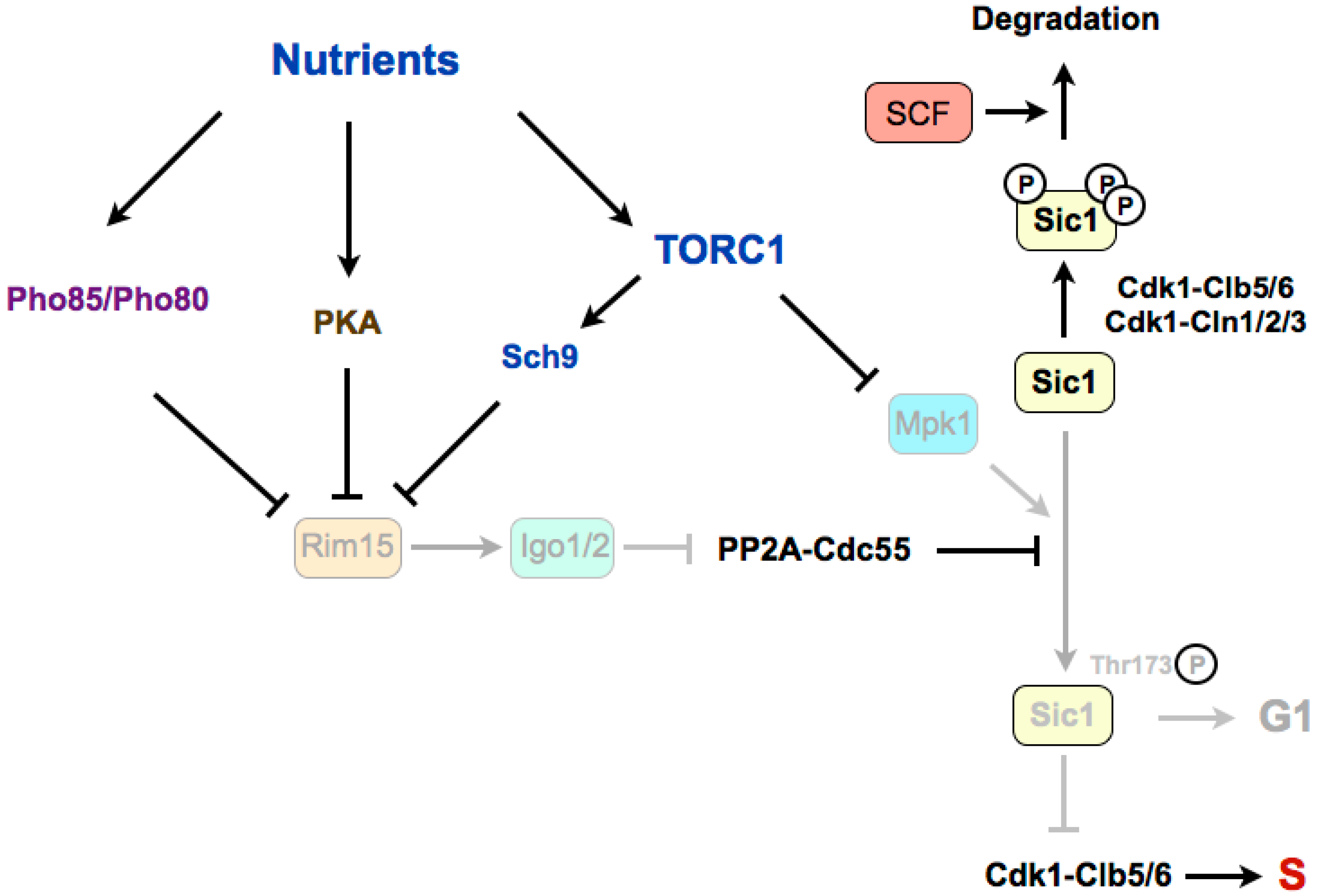
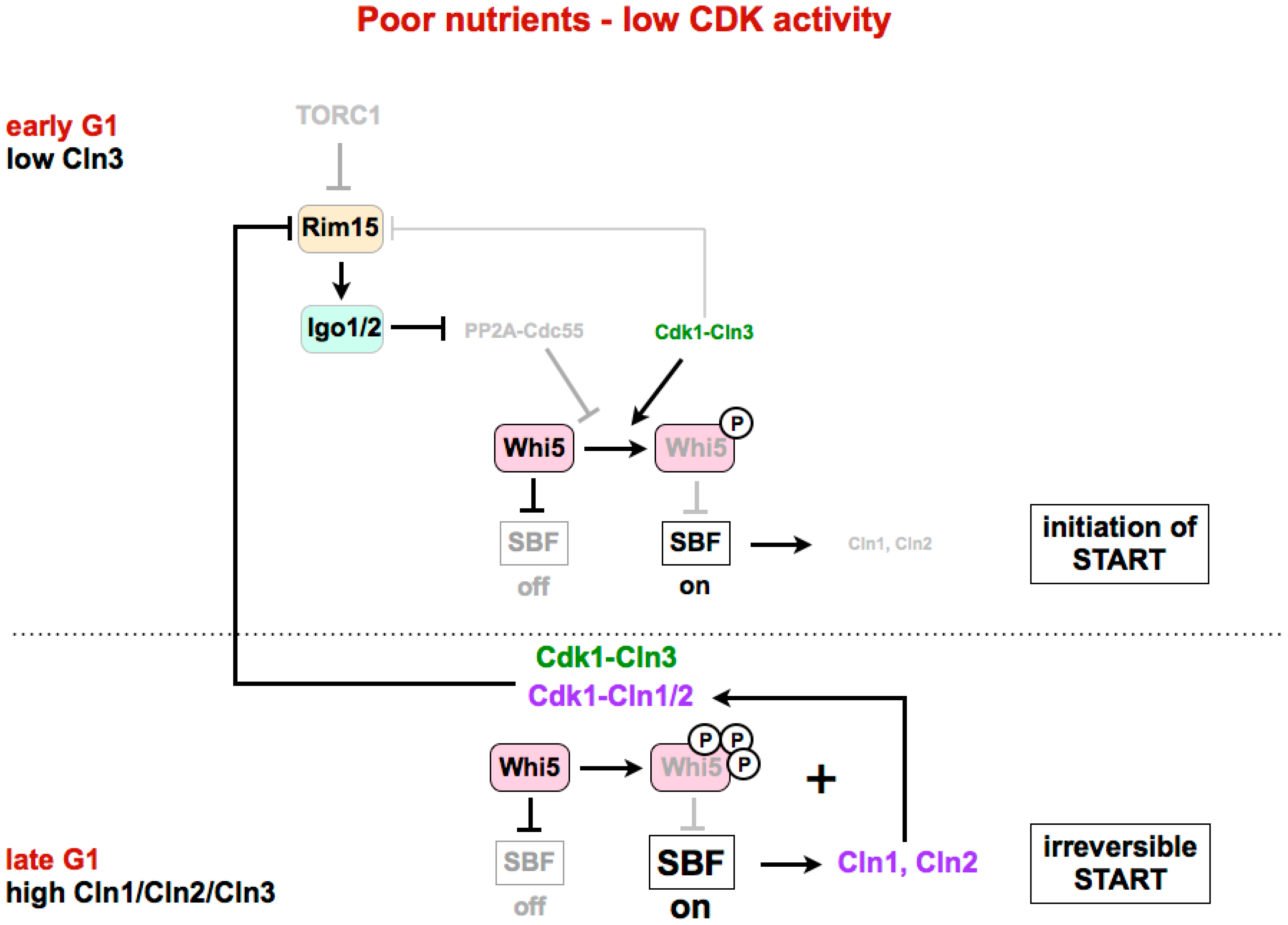
4. Regulation of G1/S in Budding Yeast
4.1. Stabilization of Sic1
4.2. Inactivation of Whi5
5. TORC1–Greatwall–Endosulfine in Quiescence in Budding and Fission Yeast
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Nurse, P.; Thuriaux, P. Controls over the timing of DNA replication during the cell cycle of fission yeast. Exp. Cell Res. 1977, 107, 365–375. [Google Scholar] [CrossRef]
- Johnston, G.C.; Pringle, J.R.; Hartwell, L.H. Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp. Cell Res. 1977, 105, 79–98. [Google Scholar] [CrossRef]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Fantes, P.; Nurse, P. Control of cell size at division in fission yeast by a growth-modulated size control over nuclear division. Exp. Cell. Res. 1977, 107, 377–386. [Google Scholar] [CrossRef]
- Weisman, R. Target of rapamycin (TOR) regulates growth in response to nutritional signals. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Alvarez, B.; Moreno, S. Fission yeast Tor2 promotes cell growth and represses cell differentiation. J. Cell Sci. 2006, 119, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Otsubo, Y.; Urano, J.; Tamanoi, F.; Yamamoto, M. Loss of the TOR kinase Tor2 mimics nitrogen starvation and activates the sexual development pathway in fission yeast. Mol. Cell Biol. 2007, 27, 3154–3164. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Hatanaka, M.; Nagao, K.; Nakaseko, Y.; Kanoh, J.; Kokubu, A.; Ebe, M.; Yanagida, M. Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 2007, 12, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular basis of the rapamycin insensitivity of Target of Rapamycin Complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Eltschinger, S.; Loewith, R. TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 2015, 26, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kunz, J.; Hall, M.N. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of ceramide biosynthesis by TOR complex 2. Cell. Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Filipuzzi, I.; Stahl, M.; Helliwell, S.B.; Studer, C.; Hoepfner, D.; Seeber, A.; Loewith, R.; Movva, N.R.; Gasser, S.M. TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol. Cell 2013, 51, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Morigasaki, S.; Tatebe, H.; Tamanoi, F.; Shiozaki, K. Fission yeast TOR complex 2 activates the AGC-family Gad8 kinase essential for stress resistance and cell cycle control. Cell Cycle 2007, 7, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Schonbrun, M.; Laor, D.; López-Maury, L.; Bähler, J.; Kupiec, M.; Weisman, R. TOR complex 2 controls gene silencing, telomere length maintenance, and survival under DNA-damaging conditions. Mol. Cell Biol. 2009, 29, 4584–4594. [Google Scholar] [CrossRef] [PubMed]
- Schonbrun, M.; Kolesnikov, M.; Kupiec, M.; Weisman, R. TORC2 is required to maintain genome stability during S phase in fission yeast. J. Biol. Chem. 2013, 288, 19649–19660. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Kirkham, S.; Halova, L.; Atkin, J.; Franz-Wachtel, M.; Cobley, D.; Krug, K.; Maček, B.; Mulvihill, D.P.; Petersen, J. TOR complex 2 localises to the cytokinetic actomyosin ring and controls the fidelity of cytokinesis. J. Cell Sci. 2016, 129, 2613–2624. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Fujita, M.; Culley, B.M.; Apolinario, E.; Yamamoto, M.; Maundrell, K.; Hoffman, C.S. Sck1, a high copy number suppressor of defects in the cAMP-dependent protein kinase pathway in fission yeast, encodes a protein homologous to the Saccharomyces cerevisiae Sch9 kinase. Genetics 1995, 140, 457–467. [Google Scholar] [PubMed]
- Fujita, M.; Yamamoto, M. S. pombe sck2+, a second homologue of S. cerevisiae SCH9 in fission yeast, encodes a putative protein kinase closely related to PKA in function. Curr. Genet. 1998, 33, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Fujioka, Y.; Suzuki, N.N.; Inagaki, F.; Wullschleger, S.; Loewith, R.; Hall, M.N.; Ohsumi, Y. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol. Cell Biol. 2005, 25, 7239–7248. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Otsubo, Y.; Yamashita, A.; Sato, T.; Yamamoto, M.; Tamanoi, F. Psk1, an AGC kinase family member in fission yeast, is directly phosphorylated and controlled by TORC1 and functions as S6 kinase. J. Cell Sci. 2012, 125, 5840–5849. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Lorberg, A. TOR regulation of AGC kinases in yeast and mammals. Biochem. J. 2008, 410, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Stern, B.; Nurse, P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996, 12, 345–350. [Google Scholar] [CrossRef]
- Coudreuse, D.; Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK substrate phosphorylation and ordering the cell cycle. Cell 2016, 167, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, M.; Touati, S.A.; Kataria, M.; Jones, A.; Snijders, A.P.; Uhlmann, F. PP2A(Cdc55) phosphatase imposes ordered cell-cycle phosphorylation by opposing threonine phosphorylation. Mol. Cell 2017, 65, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kamenz, J.; Ferrell, J.E. The temporal ordering of cell-cycle phosphorylation. Mol. Cell 2017, 65, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Cundell, M.J.; Bastos, R.N.; Zhang, T.; Holder, J.; Gruneberg, U.; Novak, B.; Barr, F.A. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol. Cell 2013, 52, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Cundell, M.J.; Hutter, L.H.; Nunes Bastos, R.; Poser, E.; Holder, J.; Mohammed, S.; Novak, B.; Barr, F.A. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J. Cell Biol. 2016, 214, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Sananes, M.R.; Kapuy, O.; Hunt, T.; Novak, B. Switches and latches: A biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Grallert, A.; Boke, E.; Hagting, A.; Hodgson, B.; Connolly, Y.; Griffiths, J.R.; Smith, D.L.; Pines, J.; Hagan, I.M. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 2015, 517, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Hunt, T. Protein phosphatases and their regulation in the control of mitosis. EMBO Rep. 2012, 13, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fleming, S.L.; Williams, B.; Williams, E.V.; Li, Z.; Somma, P.; Rieder, C.L.; Goldberg, M.L. Greatwall kinase: A nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J. Cell Biol. 2004, 164, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, Y.; Li, Z.; Galas, S.; Goldberg, M.L. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus. egg extracts. Mol. Cell 2006, 22, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Ayachi, A.; Labbé, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Voets, E.; Wolthuis, R.M. MASTL is the human orthologue of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 2010, 9, 3591–3601. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbé, J.C.; Lorca, T.; Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Fernández, M.; Sánchez-Martínez, R.; Sanz-Castillo, B.; Gan, P.P.; Sanz-Flores, M.; Trakala, M.; Ruiz-Torres, M.; Lorca, T.; Castro, A.; Malumbres, M. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc. Natl. Acad. Sci. USA 2013, 110, 17374–17379. [Google Scholar] [CrossRef] [PubMed]
- Vidan, S.; Mitchell, A.P. Stimulation of yeast meiotic gene expression by the glucose-repressible protein kinase Rim15p. Mol. Cell Biol. 1997, 17, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Reinders, A.; Bürckert, N.; Boller, T.; Wiemken, A.; De Virgilio, C. Saccharomyces cerevisiae cAMP-dependent protein kinase controls entry into stationary phase through the Rim15p protein kinase. Genes Dev. 1998, 12, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, I.; Dubouloz, F.; Cameroni, E.; Wanke, V.; Roosen, J.; Winderickx, J.; De Virgilio, C. TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol. Cell 2003, 12, 1607–1613. [Google Scholar] [CrossRef]
- Talarek, N.; Cameroni, E.; Jaquenoud, M.; Luo, X.; Bontron, S.; Lippman, S.; Devgan, G.; Snyder, M.; Broach, J.R.; De Virgilio, C. Initiation of the TORC1-regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5’-3’ mRNA decay pathway. Mol. Cell 2010, 38, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Chica, N.; Rozalen, A.E.; Perez-Hidalgo, L.; Rubio, A.; Novak, B.; Moreno, S. Nutritional control of cell size by the Greatwall-Endosulfine-PP2A·B55 pathway. Curr. Biol. 2016, 26, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Uritani, M.; Hidaka, H.; Hotta, Y.; Ueno, M.; Ushimaru, T.; Toda, T. Fission yeast Tor2 links nitrogen signals to cell proliferation and acts downstream of the Rheb GTPase. Genes Cells 2006, 11, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70S6K/p85S6K gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [PubMed]
- Montagne, J.; Stewart, M.J.; Stocker, H.; Hafen, E.; Kozma, S.C.; Thomas, G. Drosophila S6 kinase: A regulator of cell size. Science 1999, 285, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Rallis, C.; López-Maury, L.; Georgescu, T.; Pancaldi, V.; Bähler, J. Systematic screen for mutants resistant to TORC1 inhibition in fission yeast reveals genes involved in cellular ageing and growth. Biol. Open 2014, 3, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Weston, L.; Greenwood, J.; Nurse, P. Genome-wide screen for cell growth regulators in fission yeast. J. Cell Sci. 2017, 130, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Torreira, E.; Louro, J.A.; Pazos, I.; González-Polo, N.; Gil-Carton, D.; Duran, A.G.; Tosi, S.; Gallego, O.; Calvo, O.; Fernández-Tornero, C. The dynamic assembly of distinct RNA polymerase I complexes modulates rDNA transcription. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Daga, R.R.; Jimenez, J. Translational control of the Cdc25 cell cycle phosphatase: A molecular mechanism coupling mitosis to cell growth. J. Cell Sci. 1999, 112, 3137–3146. [Google Scholar] [PubMed]
- Keifenheim, D.; Sun, X.M.; D’Souza, E.; Ohira, M.J.; Magner, M.; Mayhew, M.B.; Marguerat, S.; Rhind, N. Size-dependent expression of the mitotic activator Cdc25 suggests a mechanism of size control in fission yeast. Curr. Biol. 2017, 27, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Ohkura, H.; Yanagida, M. Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell 1990, 63, 405–415. [Google Scholar] [CrossRef]
- Kinoshita, N.; Yamano, H.; Niwa, H.; Yoshida, T.; Yanagida, M. Negative regulation of mitosis by the fission yeast protein phosphatase Ppa2. Genes Dev. 1993, 7, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Nemoto, T.; Nabeshima, K.; Kondoh, H.; Niwa, H.; Yanagida, M. The regulatory subunits of fission yeast protein phosphatase 2A (PP2A) affect cell morphogenesis, cell wall synthesis and cytokinesis. Genes Cells 1996, 1, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M. The molecular control mechanisms of meiosis in fission yeast. Trends Biochem. Sci. 1996, 21, 18–22. [Google Scholar] [CrossRef]
- Weisman, R.; Choder, M. The fission yeast tor homolog, tor1+, is required for the response to starvation and other stresses via a conserved serine. J. Biol. Chem. 2000, 276, 7027–7032. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Nakashima, A.; Ueno, M.; Ushimaru, T.; Aiba, K.; Doi, H.; Uritani, M. Fission yeast Tor1 functions in response to various stresses including nitrogen starvation, high osmolarity, and high temperature. Curr. Genet. 2001, 39, 166–174. [Google Scholar] [PubMed]
- Szilagyi, Z.; Batta, G.; Enczi, K.; Sipiczki, M. Characterisation of two novel fork-head gene homologues of Schizosaccharomyces. pombe: Their involvement in cell cycle and sexual differentiation. Gene 2005, 348, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Yamada-Namikawa, C.; Murakami-Tonami, Y.; Yoshida, T.; Nakanishi, M.; Urano, T.; Murakami, H. Cdc2p controls the forkhead transcription factor Fkh2p by phosphorylation during sexual differentiation in fission yeast. EMBO J. 2007, 27, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Portantier, M.; Chica, N.; Nyquist-Andersen, M.; Mata, J.; Lopez-Aviles, S. A PP2A-B55-mediated crosstalk between TORC1 and TORC2 regulates the differentiation response in fission yeast. Curr. Biol. 2017, 27, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Weisman, R.; Roitburg, I.; Schonbrun, M.; Harari, R.; Kupiec, M. Opposite effects of tor1 and tor2 on nitrogen starvation responses in fission yeast. Genetics 2006, 175, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Lartigue, L.; Vigneron, S.; Gadea, G.; Gire, V.; Del Rio, M.; Soubeyran, I.; Chibon, F.; Lorca, T.; Castro, A. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Life 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Dalgaard, J.Z.; Millar, J.B.; Arumugam, P. The Rim15-Endosulfine-PP2A-Cdc55 signalling module regulates entry into gametogenesis and quiescence via distinct mechanisms in budding yeast. PLoS Genet. 2014, 10, e1004456. [Google Scholar] [CrossRef] [PubMed]
- Lengronne, A.; Schwob, E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G1. Mol. Cell 2002, 9, 1067–1078. [Google Scholar] [CrossRef]
- Zinzalla, V.; Graziola, M.; Mastriani, A.; Vanoni, M.; Alberghina, L. Rapamycin-mediated G1 arrest involves regulation of the CDK inhibitor Sic1 in Saccharomyces cerevisiae. Mol. Microbiol. 2007, 63, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Barbet, N.C.; Schneider, U.; Helliwell, S.B.; Stansfield, I.; Tuite, M.F.; Hall, M.N. TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 1996, 7, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Polymenis, M.; Schmidt, E.V. Coupling of cell division to cell growth by translational control of the G1 cyclin CLN3 in yeast. Genes Dev. 1997, 11, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Annan, R.S.; Huddleston, M.J.; Carr, S.A.; Reynard, G.; Deshaies, R.J. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 1997, 278, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.; Tang, X.; Orlicky, S.; Chen, Q.; Gertler, F.B.; Mendenhall, M.D.; Sicheri, F.; Pawson, T.; Tyers, M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 2001, 414, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Kõivomägi, M.; Valk, E.; Venta, R.; Iofik, A.; Lepiku, M.; Balog, E.R.; Rubin, S.M.; Morgan, D.O.; Loog, M. Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 2011, 480, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Torres, M.; Jaquenoud, M.; De Virgilio, C. TORC1 controls G1–S cell cycle transition in yeast via Mpk1 and the Greatwall kinase pathway. Nat. Commun. 2015, 6, 8256. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Torres, M.; Jaquenoud, M.; Péli-Gulli, M.P.; Nicastro, R.; De Virgilio, C. TORC1 coordinates the conversion of Sic1 from a target to an inhibitor of cyclin-CDK-Cks1. Cell Discov. 2017, 3, 17012. [Google Scholar] [CrossRef] [PubMed]
- Talarek, N.; Gueydon, E.; Schwob, E. Homeostatic control of START through negative feedback between Cln3-Cdk1 and Rim15/Greatwall kinase in budding yeast. eLife 2017, 6, e26233. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, E.; Ghillebert, R.; Wilms, T.; Winderickx, J. Molecular mechanisms linking the evolutionary conserved TORC1-Sch9 nutrient signalling branch to lifespan regulation in Saccharomyces cerevisiae. FEMS Yeast Res. 2013, 14, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Wanke, V.; Pedruzzi, I.; Cameroni, E.; Dubouloz, F.; De Virgilio, C. Regulation of G0 entry by the Pho80-Pho85 cyclin-CDK complex. EMBO J. 2005, 24, 4271–4278. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, E.; Wanke, V.; Roosen, J.; Smets, B.; Dubouloz, F.; Pedruzzi, I.; Cameroni, E.; De Virgilio, C.; Winderickx, J. Rim15 and the crossroads of nutrient signalling pathways in Saccharomyces cerevisiae. Cell Div. 2006, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Bontron, S.; Jaquenoud, M.; Vaga, S.; Talarek, N.; Bodenmiller, B.; Aebersold, R.; De Virgilio, C. Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep. 2013, 3, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Kim, M.S.; Paik, S.M.; Choi, S.H.; Cho, B.R.; Hahn, J.S. Rim15-dependent activation of Hsf1 and Msn2/4 transcription factors by direct phosphorylation in Saccharomyces cerevisiae. FEBS Lett. 2013, 587, 3648–3655. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Talarek, N.; De Virgilio, C. Initiation of the yeast G0 program requires Igo1 and Igo2, which antagonize activation of decapping of specific nutrient-regulated mRNAs. RNA Biol. 2011, 8, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Talarek, N.; Bontron, S.; De Virgilio, C. Quantification of mRNA stability of stress-responsive yeast genes following conditional excision of open reading frames. RNA Biol. 2013, 10, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.R.; Li, Y.; Eisenstatt, J.R.; Runge, K.W. Identification of a lifespan extending mutation in the Schizosaccharomyces. pombe cyclin gene clg1+ by direct selection of long-lived mutants. PLoS ONE 2013, 8, e69084. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Kupiec, M.; Weisman, R. Gad8 protein is found in the nucleus where it interacts with the MluI Cell Cycle Box-binding factor (MBF) transcriptional complex to regulate the response to DNA replication stress. J. Biol. Chem. 2016, 291, 9371–9381. [Google Scholar] [CrossRef] [PubMed]
- Weisman, R.; Cohen, A.; Gasser, S.M. TORC2-a new player in genome stability. EMBO Mol. Med. 2014, 6, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, J.; Elia, A.; Carroll, V.A.; Moumen, A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2015, 6, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Maruki, Y.; Imamura, Y.; Kondo, C.; Kawamata, T.; Kawanishi, I.; Takata, H.; Matsuura, A.; Lee, K.S.; Kikkawa, U.; et al. The yeast Tor signaling pathway is involved in G2/M transition via polo-kinase. PLoS ONE 2008, 3, e2223. [Google Scholar] [CrossRef] [PubMed]
- Zacharek, S.J.; Xiong, Y.; Shumway, S.D. Negative regulation of Tsc1-Tsc2 by mammalian D-type cyclins. Cancer Res. 2005, 65, 11354–11360. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cdk4/6 inhibitors resTORe therapeutic sensitivity in HER2+ breast cancer. Cancer Cell 2016, 29, 243–244. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| S. cerevisiae | S. pombe | Mammals | |
|---|---|---|---|
| TORC1 | Tor1 or Tor2 | Tor2 | mTOR |
| Kog1 (Raptor) | Mip1 (Raptor) | Raptor | |
| — | — | Deptor | |
| Lst8 | Wat1 | mLst8 | |
| TORC2 | Tor2 | Tor1 | mTOR |
| Avo3 (Rictor) | Ste20 (Rictor) | Rictor | |
| Avo1 | Sin1 | mSin1 | |
| Bit61 | Bit61 | Protor | |
| — | — | Deptor | |
| Lst8 | Wat1 | mLst8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Hidalgo, L.; Moreno, S. Coupling TOR to the Cell Cycle by the Greatwall–Endosulfine–PP2A-B55 Pathway. Biomolecules 2017, 7, 59. https://doi.org/10.3390/biom7030059
Pérez-Hidalgo L, Moreno S. Coupling TOR to the Cell Cycle by the Greatwall–Endosulfine–PP2A-B55 Pathway. Biomolecules. 2017; 7(3):59. https://doi.org/10.3390/biom7030059
Chicago/Turabian StylePérez-Hidalgo, Livia, and Sergio Moreno. 2017. "Coupling TOR to the Cell Cycle by the Greatwall–Endosulfine–PP2A-B55 Pathway" Biomolecules 7, no. 3: 59. https://doi.org/10.3390/biom7030059
APA StylePérez-Hidalgo, L., & Moreno, S. (2017). Coupling TOR to the Cell Cycle by the Greatwall–Endosulfine–PP2A-B55 Pathway. Biomolecules, 7(3), 59. https://doi.org/10.3390/biom7030059