
CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles

 , ,
, ,  and
and 
{kind=link}
Abstract
:1. Introduction
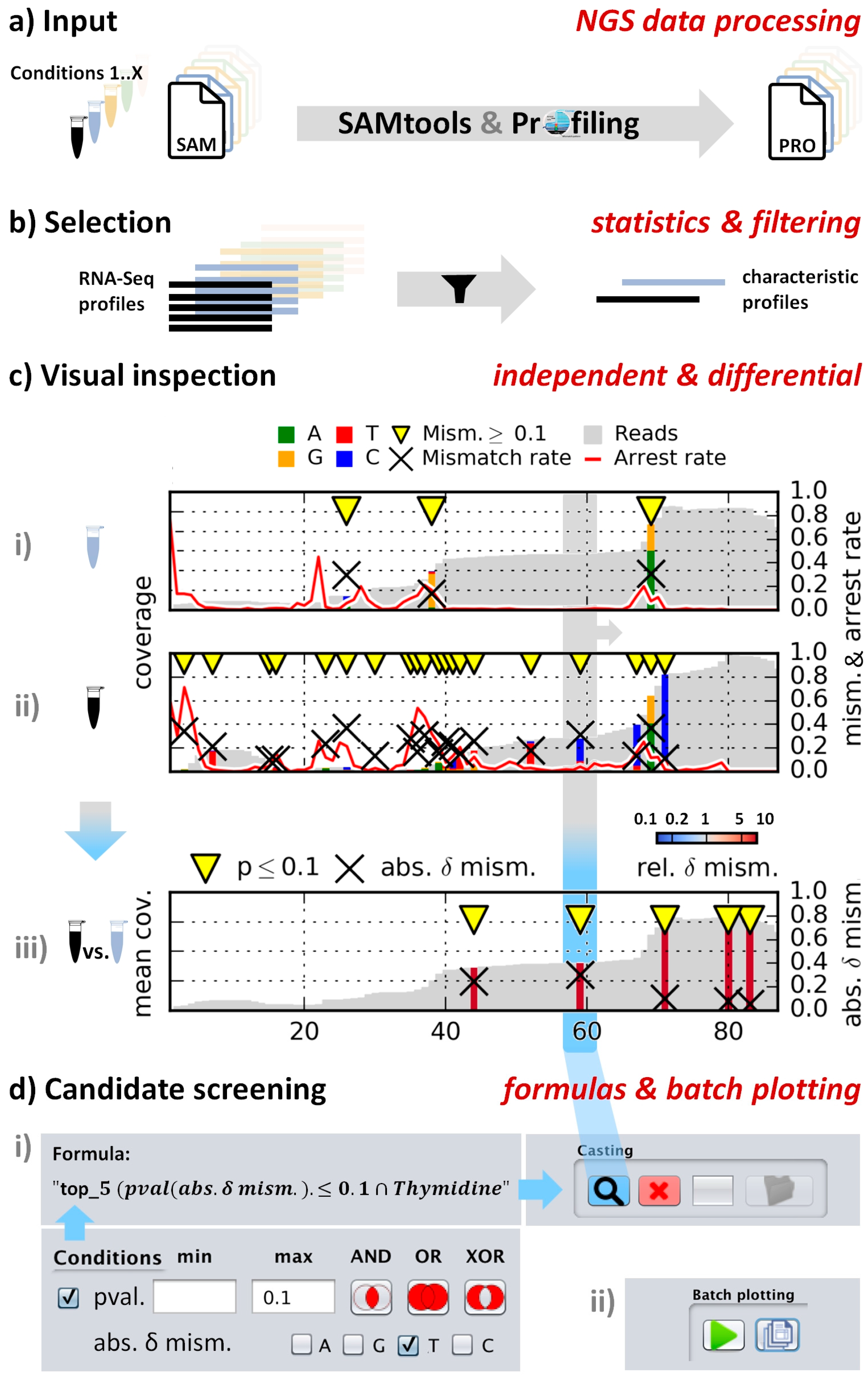
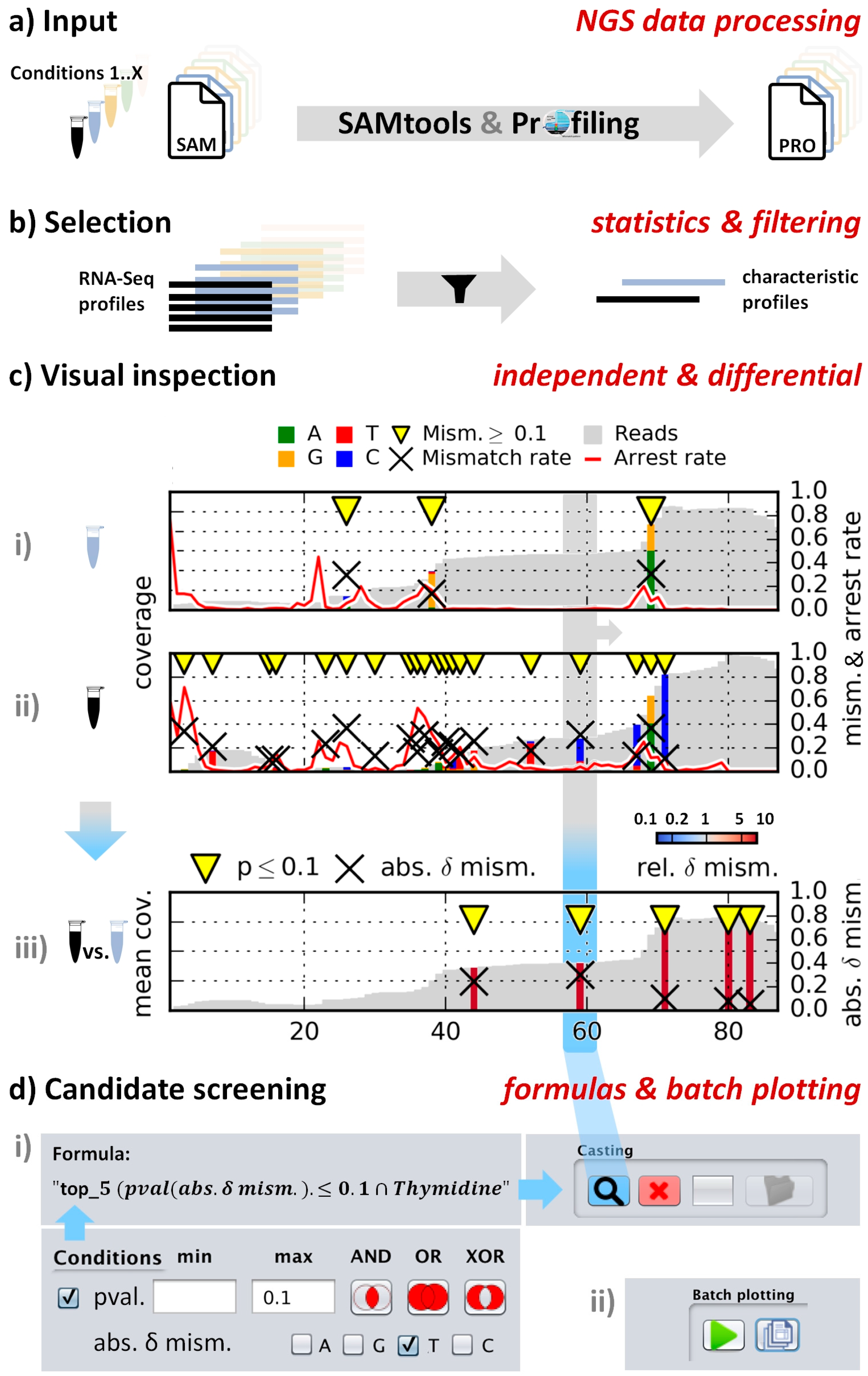
2. Results
3. Discussion
4. Materials and Methods
4.1. Implementation
4.2. Workflow
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Motorin, Y.; Helm, M. tRNA stabilization by modified nucleotides. Biochemistry 2010, 49, 4934–4944. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhao, B.S.; He, C. Nucleic acid modifications in regulation of gene expression. Cell Chem. Biol. 2016, 23, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Jaffrey, S.R.; Pan, T.; Rechavi, G.; Suzuki, T. RNA modifications: What have we learned and where are we headed? Nat. Rev. Genet. 2016, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Spenkuch, F.; Motorin, Y.; Helm, M. Pseudouridine: Still mysterious, but never a fake (uridine)! RNA Biol. 2014, 11, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dalpke, A.; Helm, M. RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol. 2012, 9, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. Modomics: A database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Alfonzo, J.D. Posttranscriptional RNA Modifications: Playing metabolic games in a cell’s chemical legoland. Chem. Biol. 2014, 21, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Kellner, S.; Neumann, J.; Rosenkranz, D.; Lebedeva, S.; Ketting, R.F.; Zischler, H.; Schneider, D.; Helm, M. Profiling of RNA modifications by multiplexed stable isotope labelling. Chem. Commun. 2014, 50, 3516–3518. [Google Scholar] [CrossRef] [PubMed]
- Lempereur, L.; Nicoloso, M.; Riehl, N.; Ehresmann, C.; Ehresmann, B.; Bachellerie, J.P. Conformation of yeast 18S rRNA. Direct chemical probing of the 5’ domain in ribosomal subunits and in deproteinized RNA by reverse transcriptase mapping of dimethyl sulfate-accessible. Nucleic Acids Res. 1985, 13, 8339–8357. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Ebhardt, H.A.; Tsang, H.H.; Dai, D.C.; Liu, Y.; Bostan, B.; Fahlman, R.P. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009, 37, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Findeiss, S.; Langenberger, D.; Stadler, P.F.; Hoffmann, S. Traces of post-transcriptional RNA modifications in deep sequencing data. Biol. Chem. 2011, 392, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.S. HAMR: High-throughput annotation of modified ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Behm-Ansmant, I.; Helm, M.; Motorin, Y. Use of specific chemical reagents for detection of modified nucleotides in RNA. J. Nucleic Acids 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; Leon-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Li, J.H.; Liu, S.; Wu, J.; Zhou, H.; Qu, L.H.; Yang, J.H. RMbase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thuring, K.; Winz, M.L.; Sharma, S.; Entian, K.D.; Wacheul, L.; Lafontaine, D.L.; Anderson, J.; et al. The reverse transcription signature of N-1-methyladenosine in RNA-seq is sequence dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Pseudo-Seq: Genome-wide detection of pseudouridine modifications in RNA. Methods Enzymol. 2015, 560, 219–245. [Google Scholar] [PubMed]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. BioTechniques 2014, 56, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Bonferroni, C. Sulle medie multiple di potenze. Bollettino dell’Unione Matematica Italiana 1950, 5, 267–270. (In Italian) [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueda, H.; Okada, S.; Sakurai, M. Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-Seq method. Nat. Protoc. 2015, 10, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The numpy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauenschild, R.; Werner, S.; Tserovski, L.; Hildebrandt, A.; Motorin, Y.; Helm, M. CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules 2016, 6, 42. https://doi.org/10.3390/biom6040042
Hauenschild R, Werner S, Tserovski L, Hildebrandt A, Motorin Y, Helm M. CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules. 2016; 6(4):42. https://doi.org/10.3390/biom6040042
Chicago/Turabian StyleHauenschild, Ralf, Stephan Werner, Lyudmil Tserovski, Andreas Hildebrandt, Yuri Motorin, and Mark Helm. 2016. "CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles" Biomolecules 6, no. 4: 42. https://doi.org/10.3390/biom6040042
APA StyleHauenschild, R., Werner, S., Tserovski, L., Hildebrandt, A., Motorin, Y., & Helm, M. (2016). CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules, 6(4), 42. https://doi.org/10.3390/biom6040042