
Absence of a Role for Phosphorylation in the Tau Pathology of Alzheimer’s Disease



Abstract
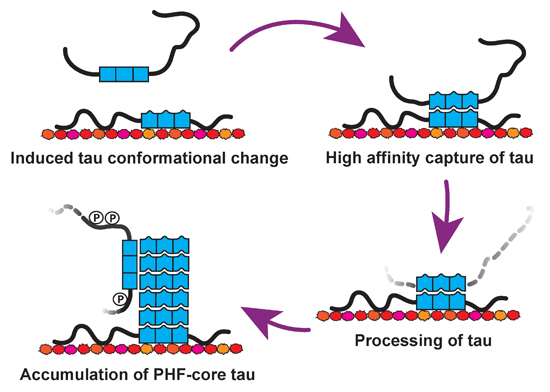
:
1. Introduction
2. Materials and Methods
3. Results
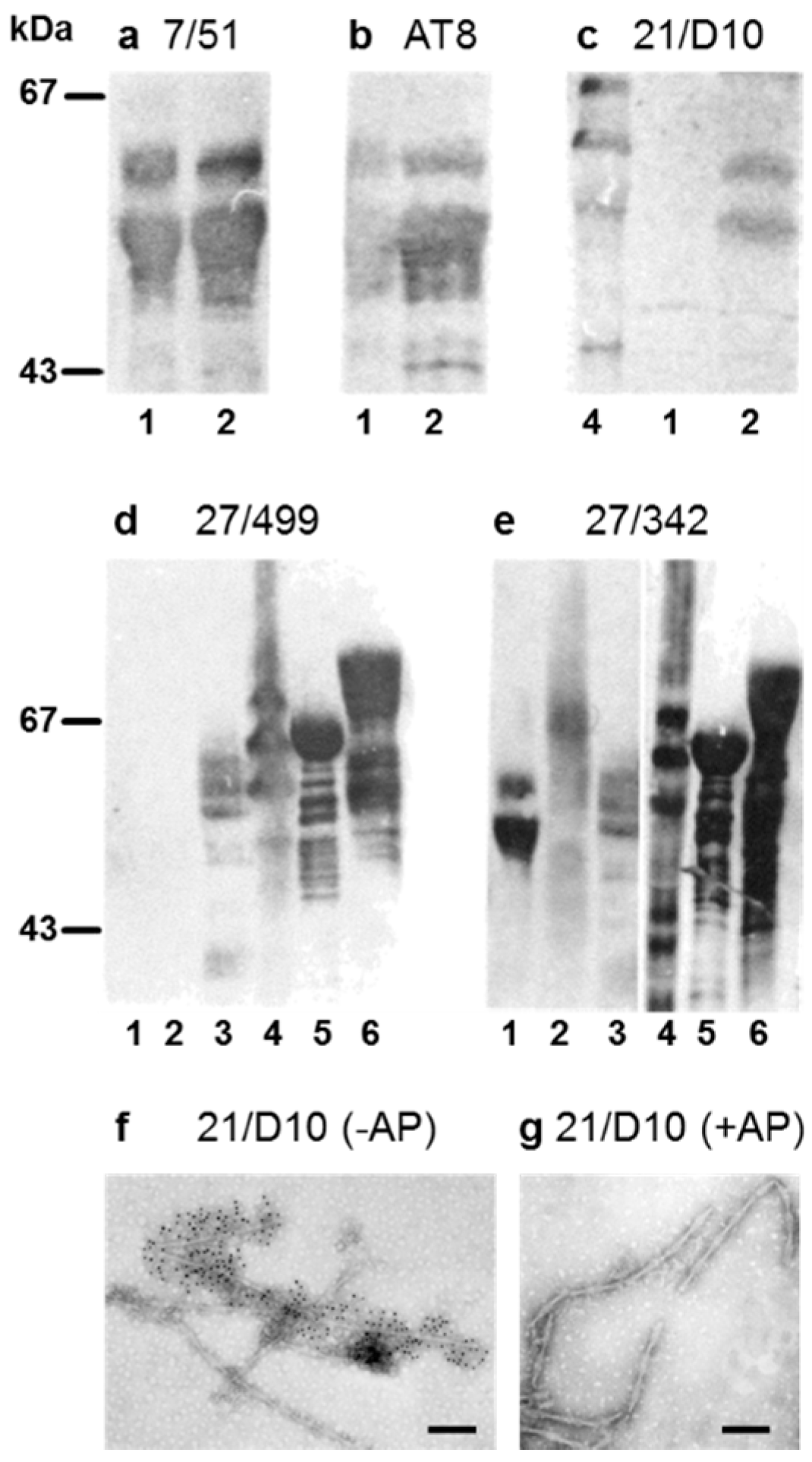
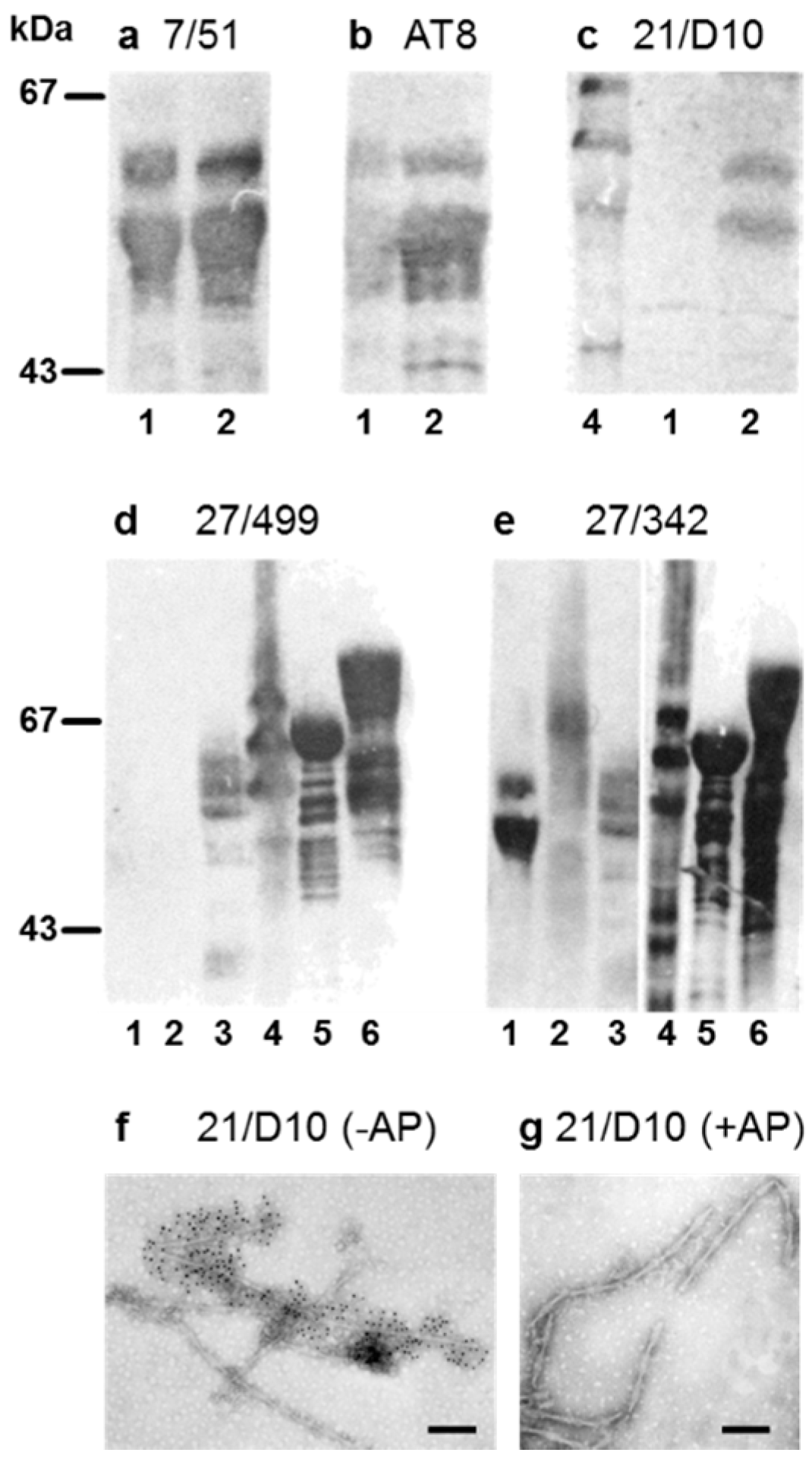
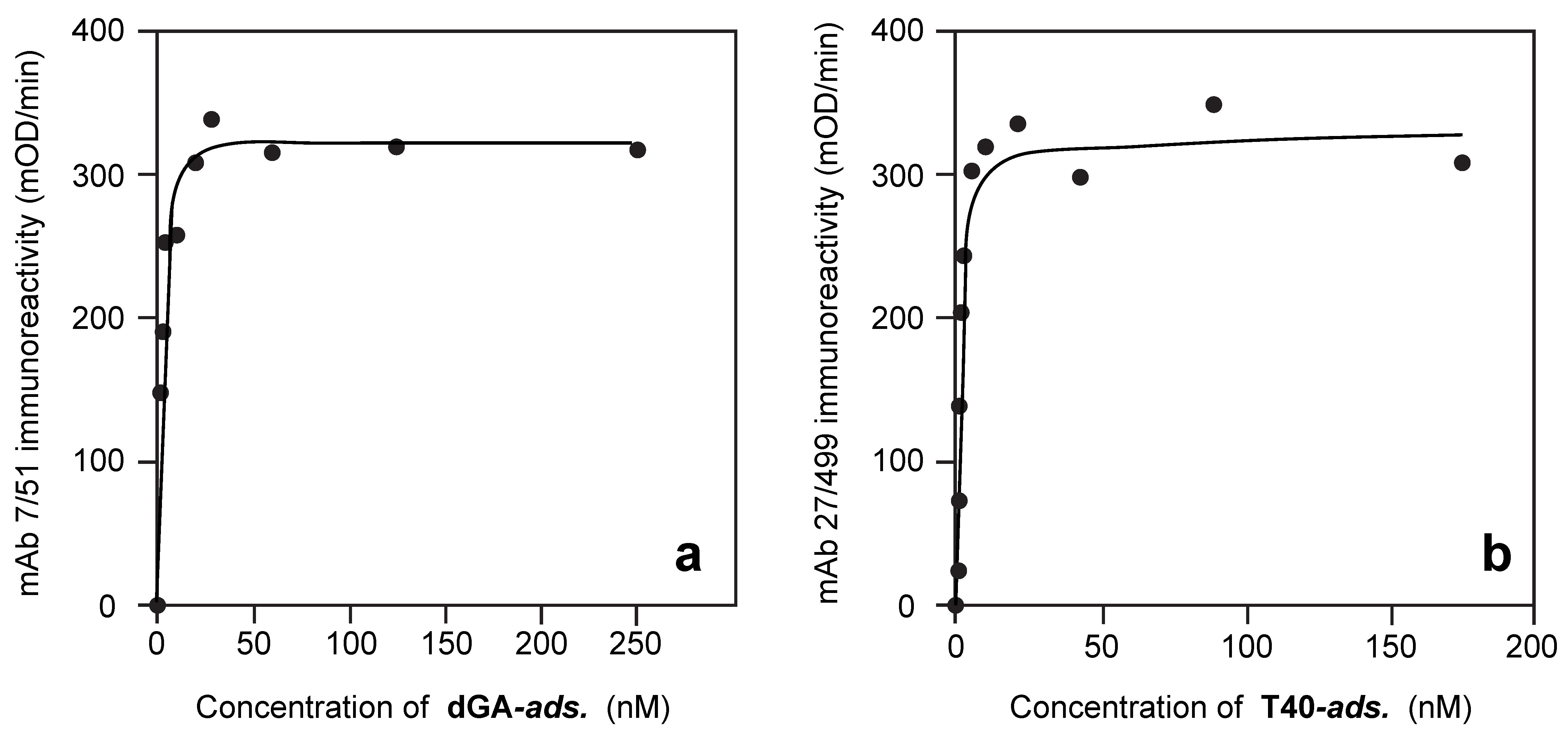
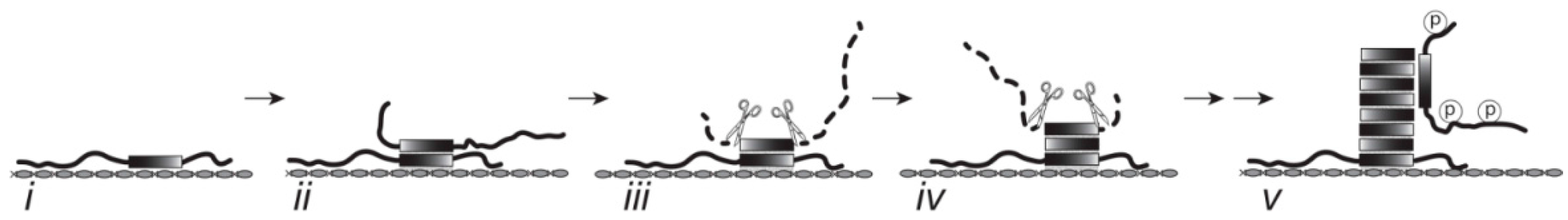
3.1. Properties of Antibodies Used to Measure Tau-Tau Binding Interactions
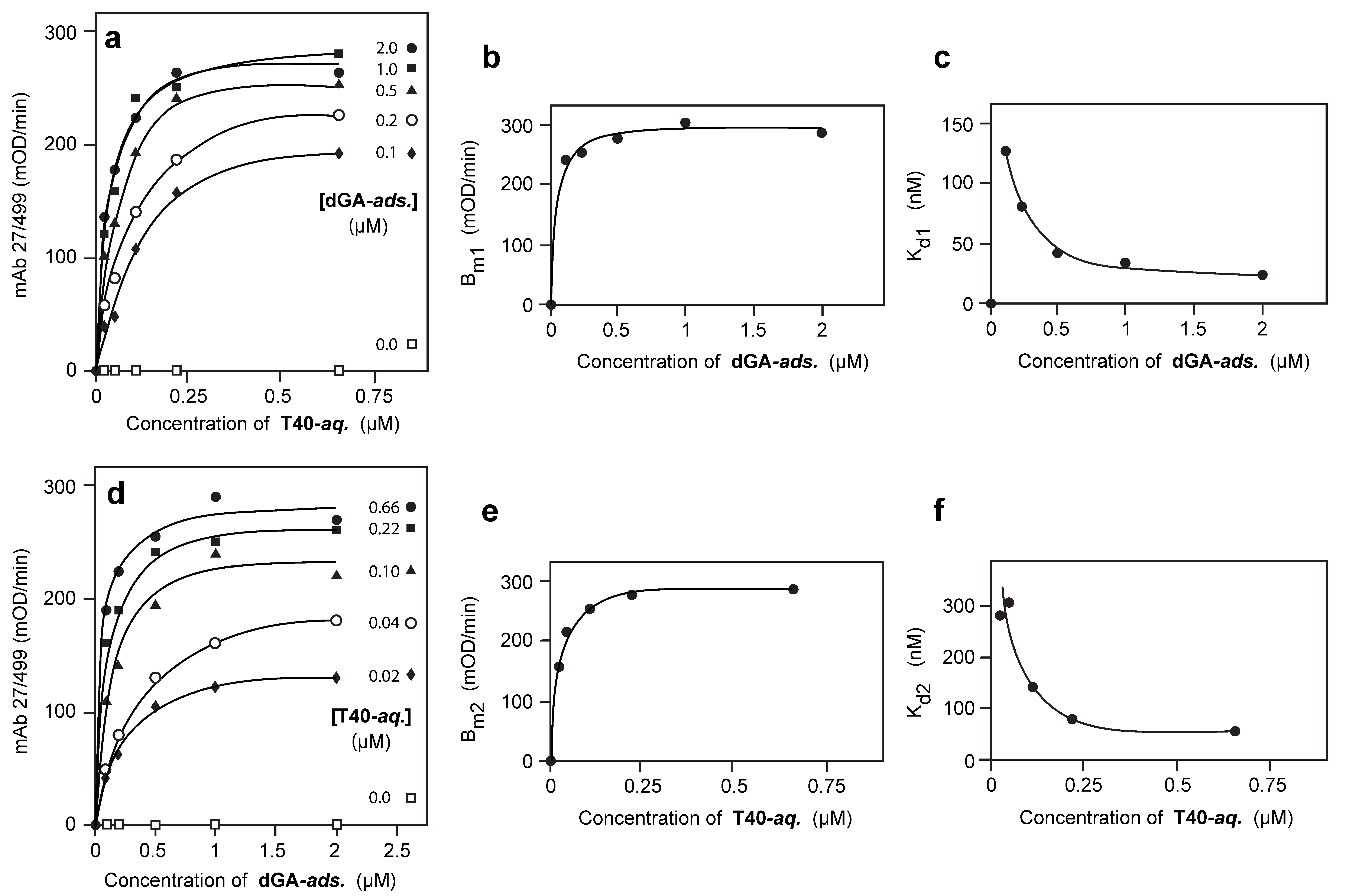
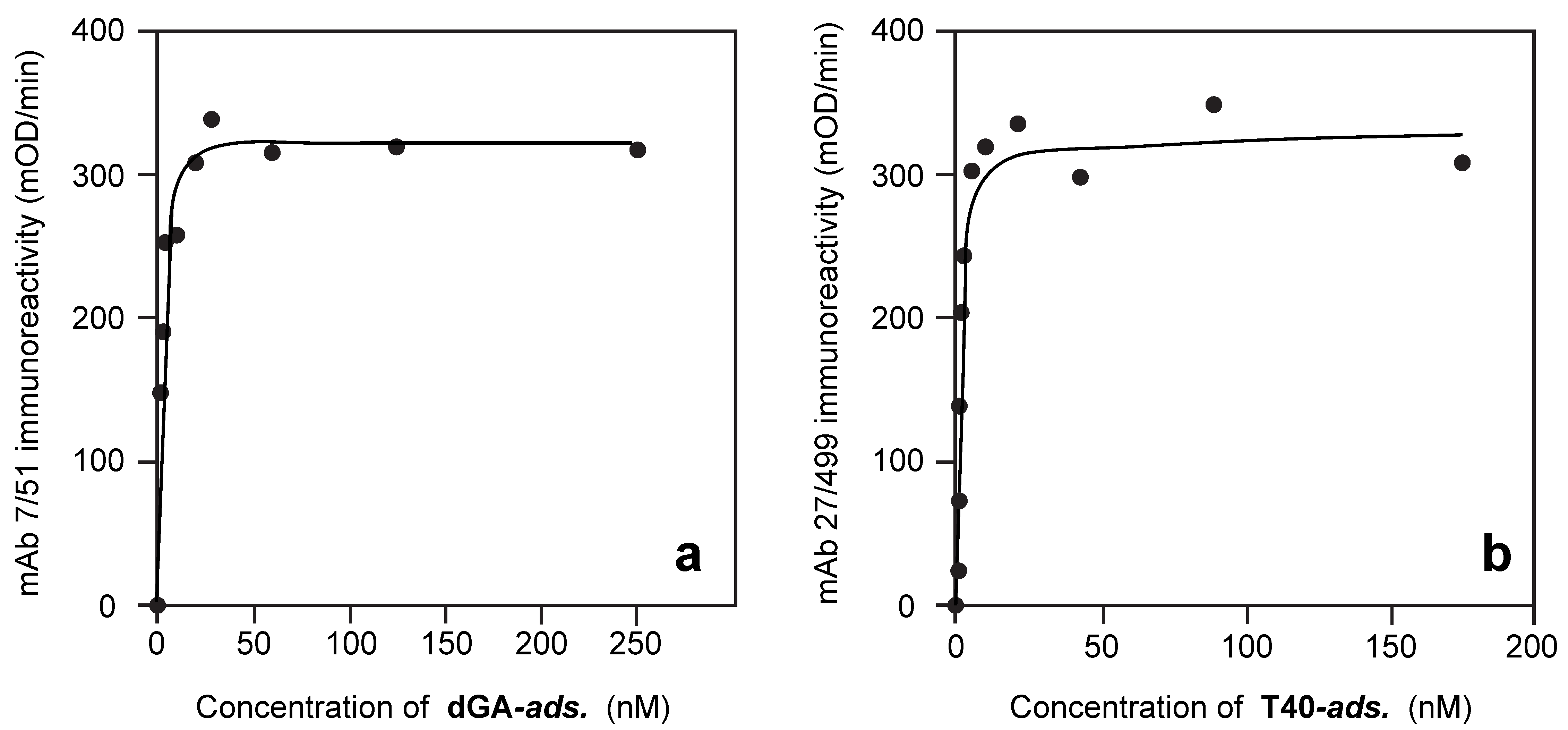
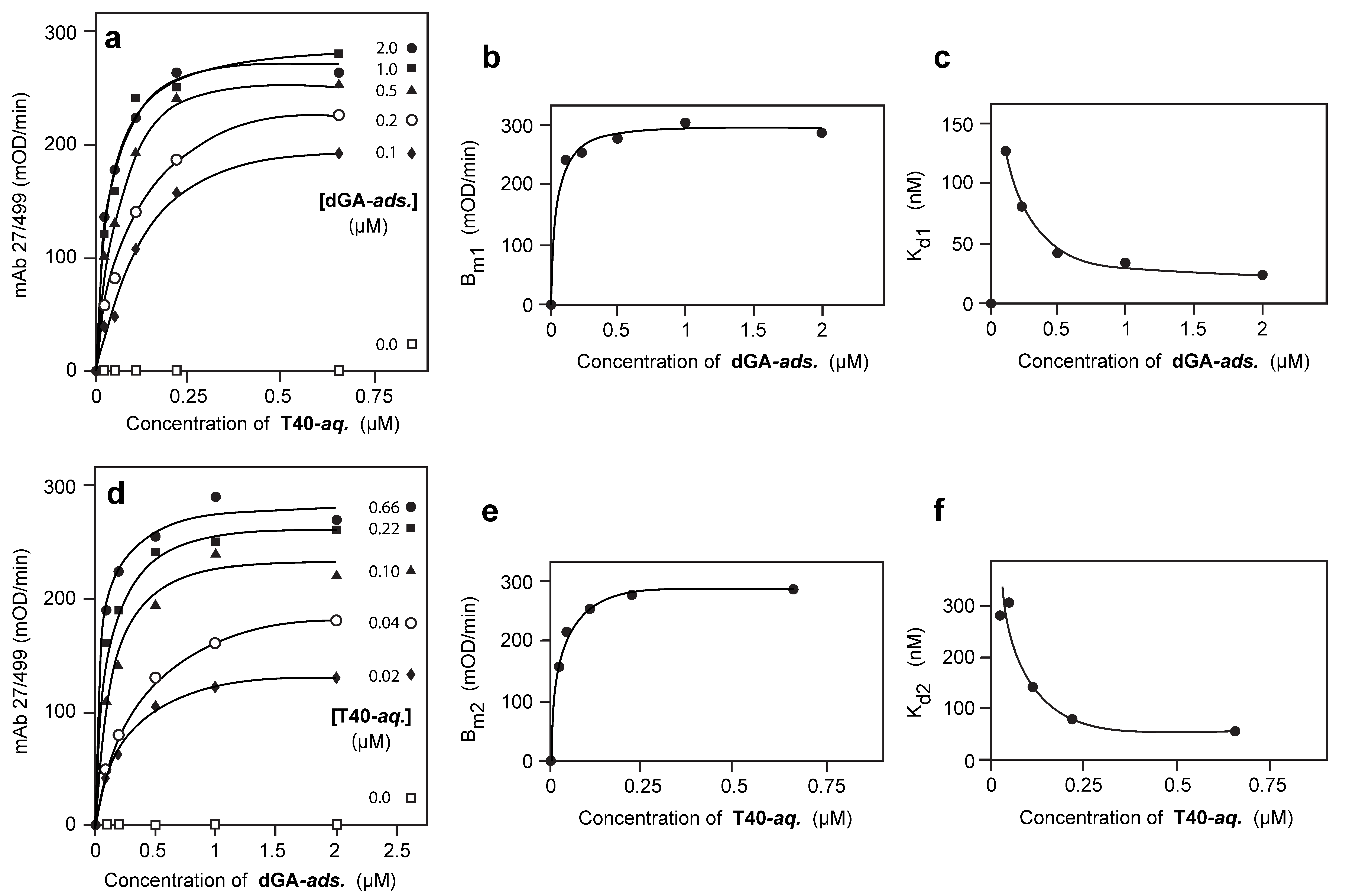
3.2. Solid-Phase Concentration-Dependent Enhancement of Tau Binding Affinity
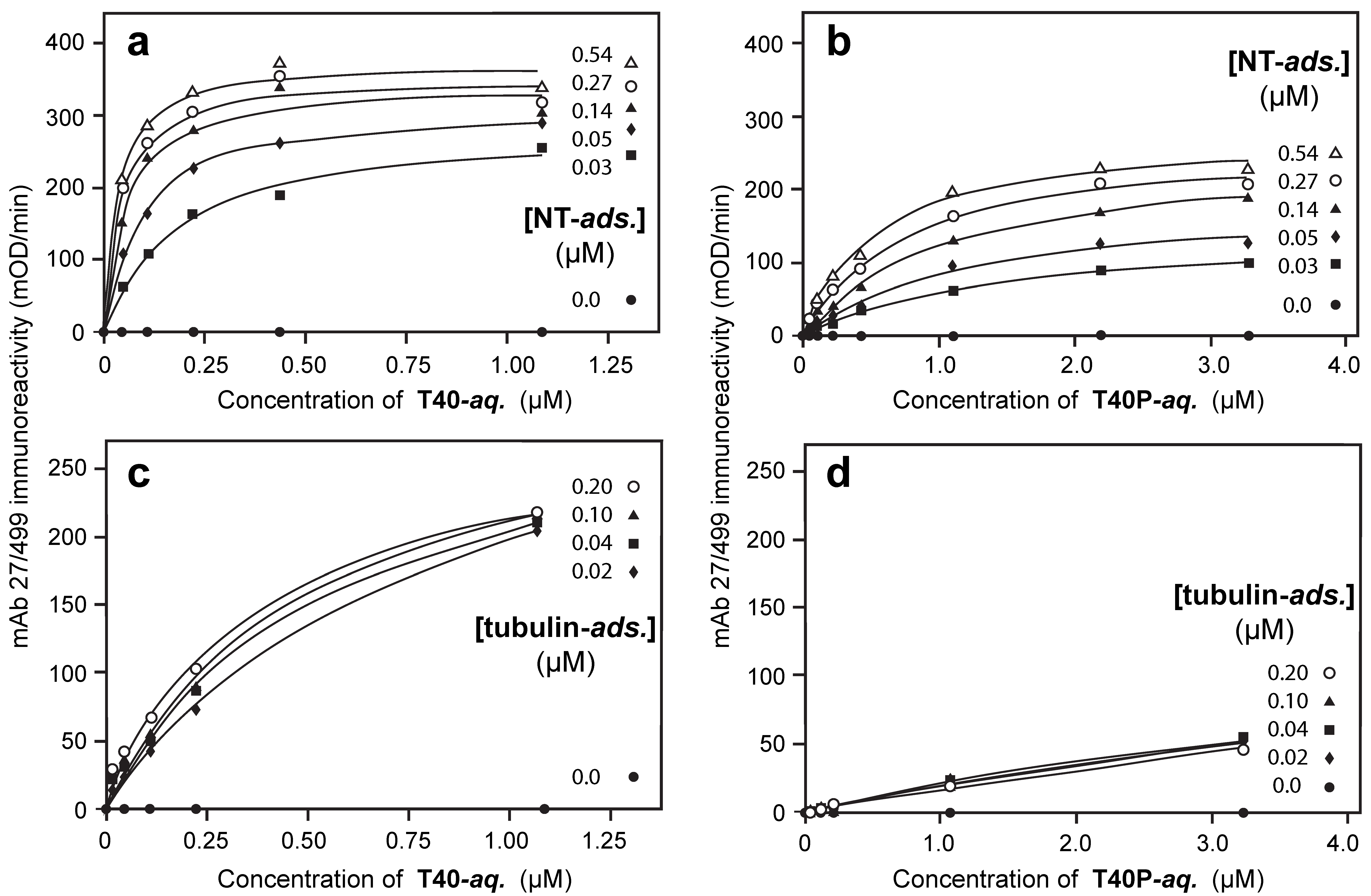
3.3. Aqueous-Phase Concentration-Dependent Enhancement of Tau Binding Affinity
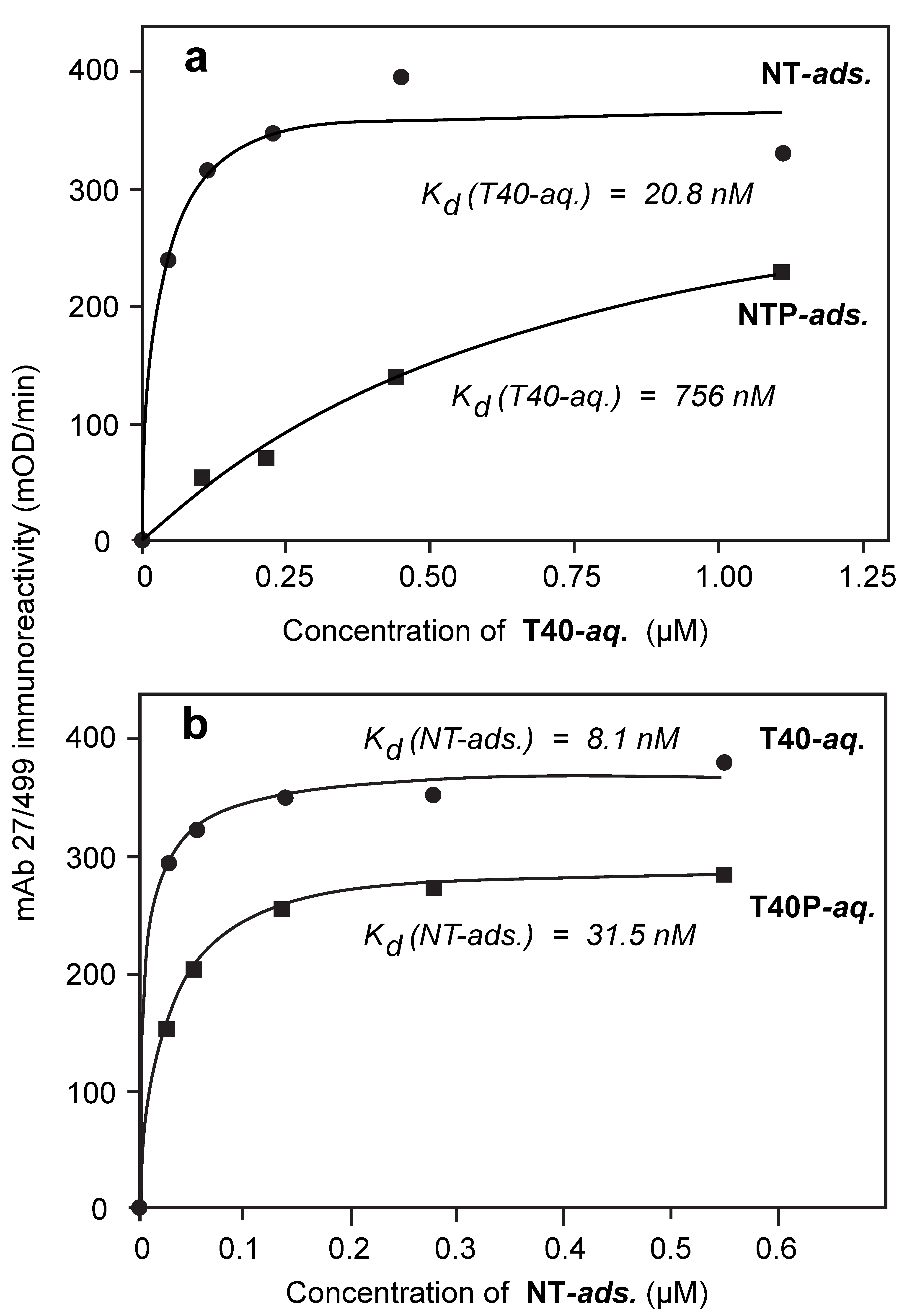
3.4. Comparison of Tau-Tau Binding Binding Affinity Constants in Aqueous and Adsorbed Phases
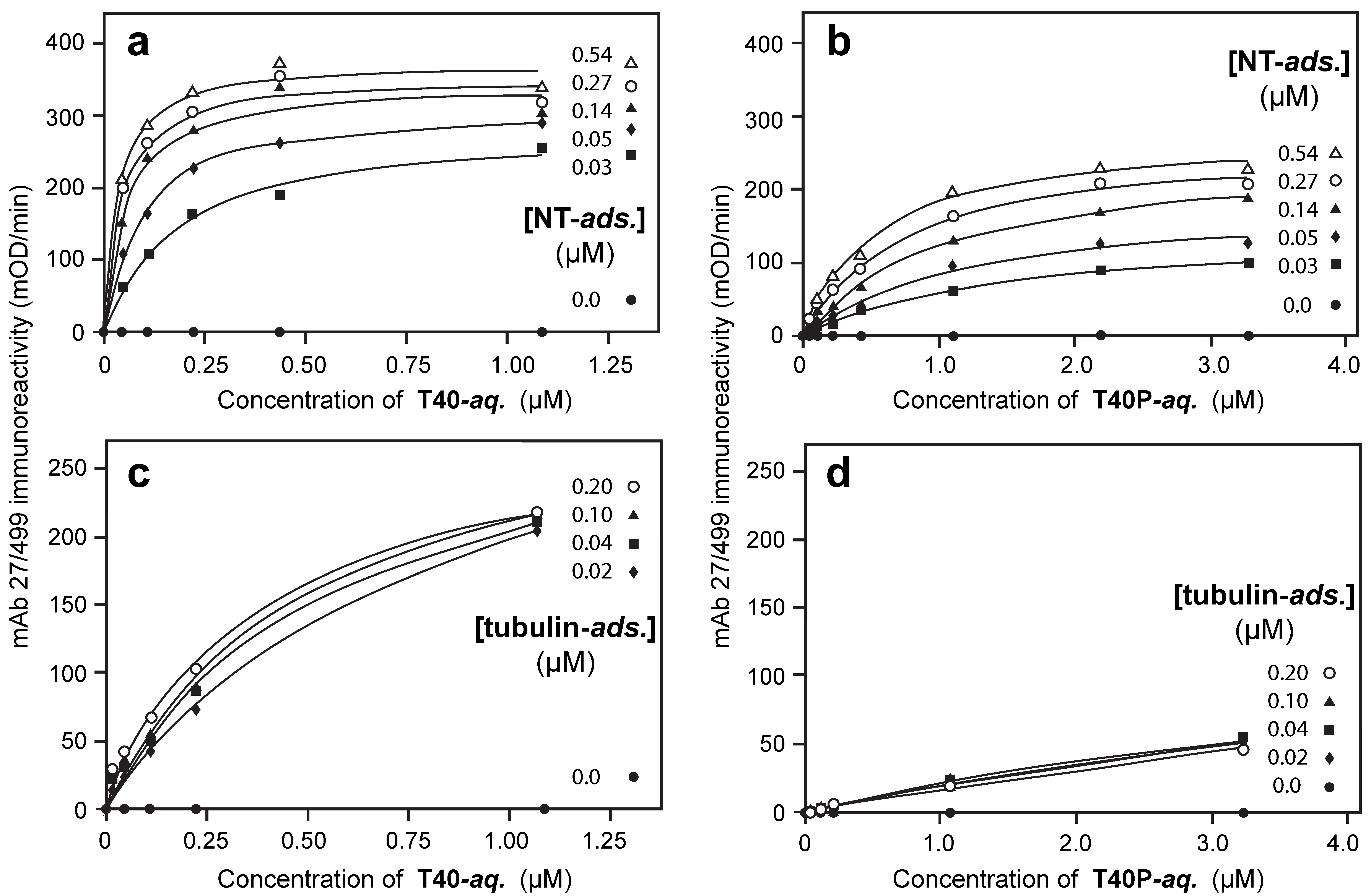
3.5. Binding Affinities in the Aqueous Phase Following (Hyper)Phosphorylation of Tau Protein
3.6. Binding of Phosphorylated or Non-Phosphorylated Tau in the Aqueous Phase to Depolymerized Tubulin
3.7. Solid-Phase Binding Constants in the Presence or Absence of Hyperphosphorylation
4. Discussion
4.1. Features of the Tau-Tau Binding Assays
4.2. The Binding Characteristics for Tau in Solid and Aqueous Phases
4.3. Impact of Phosphorylation on Binding Characteristics
4.4. Effect of Regions Outside the Repeat Domain on Binding
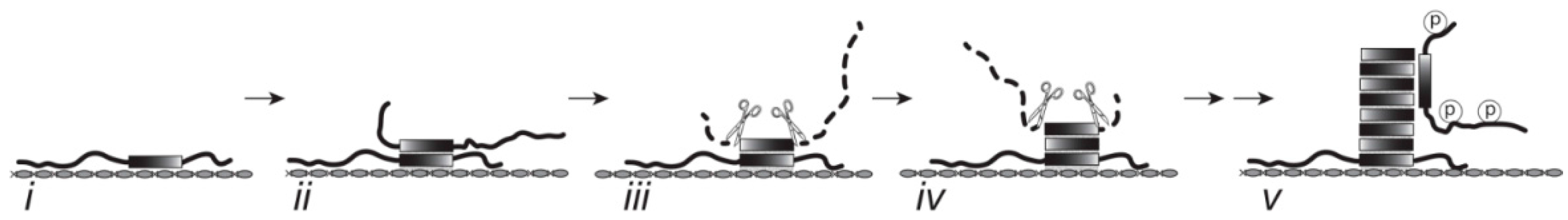
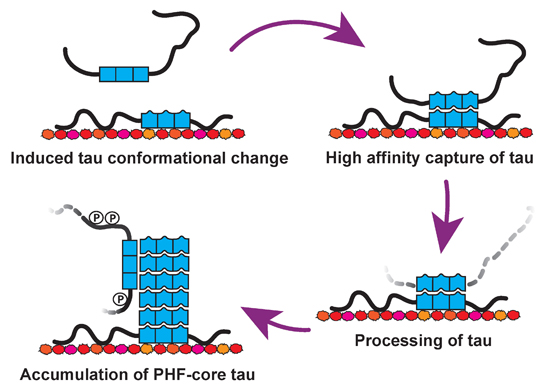
4.5. Tau-Tubulin Interactions
4.6. Therapeutic Implications
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harrington, C.R.; Mukaetova-Ladinska, E.B.; Hills, R.; Edwards, P.C.; Montejo de Garcini, E.; Novak, M.; Wischik, C.M. Measurement of distinct immunochemical presentations of tau protein in Alzheimer disease. PNAS 1991, 88, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psych. Psych. Gerich. Med. 1907, 64, 146–148. [Google Scholar]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Fodero-Tavoletti, M.; Furumoto, S.; Taylor, L.; McLean, C.; Mulligan, R.; Birchall, I.; Harada, R.; Masters, C.; Yanai, K.; Kudo, Y.; et al. Assessing THK523 selectivity for tau deposits in Alzheimer’s disease and non Alzheimer’s disease tauopathies. Alzheimers Res. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.W.; Liu, C.H.; Mocharla, V.P.; et al. F-18 T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.-Y.; Balin, B.J.; Otvos, L.J., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.-M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Alonso, A.D.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer’s disease. PNAS 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-C.; Lu, Q.; Graves, D.J. Phosphorylated tau can promote tubulin assembly. PNAS 1999, 96, 9503–9508. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Troquier, L.; Burnouf, S.; Belarbi, K.; van der Jeugd, A.; Ahmed, T.; Fernandez-Gomez, F.; Caillierez, R.; Grosjean, M.E.; Begard, S.; et al. From tau phosphorylation to tau aggregation: What about neuronal death? Biochem. Soc. Trans. 2010, 38, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.K.; Gertz, H.-J.; Xuereb, J.H.; Paykel, E.S.; Brayne, C.; Huppert, F.A.; Mukaetova-Ladinska, E.B.; Mena, R.; et al. Quantitative analysis of tau protein in paired helical filament preparations: Implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer’s disease. Neurobiol. Aging 1995, 16, 409–431. [Google Scholar] [CrossRef]
- Mukaetova-Ladinska, E.B.; Garcia-Sierra, F.; Hurt, J.; Gertz, H.J.; Xuereb, J.H.; Hills, R.; Brayne, C.; Huppert, F.A.; Paykel, E.S.; McGee, M.; et al. Staging of cytoskeletal and b-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am. J. Pathol. 2000, 157, 623–636. [Google Scholar] [CrossRef]
- Wischik, C.M.; Lai, R.Y.K.; Harrington, C.R. Modelling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. In Brain Microtubule Associated Proteins: Modifications in Disease; Avila, J., Brandt, R., Kosik, K.S., Eds.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1997; pp. 185–241. [Google Scholar]
- Novak, M.; Jakes, R.; Edwards, P.C.; Milstein, C.; Wischik, C.M. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of mAbs 423 and 7.51. PNAS 1991, 88, 5837–5841. [Google Scholar] [CrossRef] [PubMed]
- Bondareff, W.; Harrington, C.; Wischik, C.M.; Hauser, D.L.; Roth, M. Immunohistochemical staging of neurofibrillary degeneration in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1994, 53, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Bondareff, W.; Wischik, C.M.; Novak, M.; Roth, M. Sequestration of tau by granulovacuolar degeneration in Alzheimer’s disease. Am. J. Pathol. 1991, 139, 641–647. [Google Scholar] [PubMed]
- Wischik, C.M.; Novak, M.; Thøgersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer’s disease. PNAS 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Mena, R.; Edwards, P.C.; Harrington, C.R.; Mukaetova-Ladinska, E.B.; Wischik, C.M. Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 1996, 91, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Mena, R.; Edwards, P.; Pérez-Olvera, O.; Wischik, C.M. Monitoring pathological assembly of tau and b-amyloid proteins in Alzheimer’s disease. Acta Neuropathol. 1995, 89, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Novak, M.; Davison, M.; Wischik, C.M. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J. 1991, 10, 2725–2729. [Google Scholar] [PubMed]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.K.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. PNAS 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Pérez, M.; Valpuesta, J.M.; Avila, J. Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am. J. Pathol. 1997, 151, 1115–1122. [Google Scholar] [PubMed]
- Friedhoff, P.; von Bergen, M.; Mandelkow, E.-M.; Davies, P.; Mandelkow, E. A nucleated assembly of Alzheimer paired helical filaments. PNAS 1998, 95, 15712–15717. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Crowther, R.A.; Jakes, R.; Goedert, M. Alzheimer-like changes in microtubule-associated protein tau induced by sulfated glycosaminoglycans: inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J. Biol. Chem. 1997, 272, 33118–33124. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of t into filaments in the presence of heparin: the minimal sequence required for t-t interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerisation. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [PubMed]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Six, J.; Lübke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.Q.; Lee, V.M.-J. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer’s disease recapitulates phosphorylation during development. PNAS 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [PubMed]
- Biernat, J.; Mandelkow, E.-M.; Schröter, C.; Lichtenberg-Kraag, B.; Steiner, B.; Berling, B.; Meyer, H.; Mercken, M.; Vandermeeren, A.; Goedert, M.; et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992, 11, 1593–1597. [Google Scholar] [PubMed]
- Harrington, C.R.; Edwards, P.C.; Wischik, C.M. Competitive ELISA for measurement of tau proteins in Alzheimer’s disease. J. Immunol. Meth. 1990, 134, 261–271. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–170. [Google Scholar] [CrossRef]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the t in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M.-Y. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Akoury, E.; Pickhardt, M.; Gajda, M.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Mechanistic basis of phenothiazine-driven inhibition of tau aggregation. Angew. Chem. Int. Ed. 2013, 52, 3511–3515. [Google Scholar] [CrossRef] [PubMed]
- Huvent, I.; Kamah, A.; Cantrelle, F.-X.; Barois, N.; Slomianny, C.; Smet-Nocca, C.; Landrieu, I.; Lippens, G. A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge. Biochem. Biophys. Res. Commun. 2014, 445, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Olesen, O.F.; Jakes, R.; Goedert, M. The microtubule binding repeats of tau protein assemble into filaments like those found in Alzheimer’s disease. FEBS Lett. 1992, 309, 199–202. [Google Scholar] [CrossRef]
- Lai, R.Y.K.; Gertz, H.-J.; Wischik, D.J.; Xuereb, J.H.; Mukaetova-Ladinska, E.B.; Harrington, C.R.; Edwards, P.C.; Mena, R.; Paykel, E.S.; Brayne, C.; et al. Examination of phosphorylated tau protein as a PHF-precursor at early stage Alzheimer’s disease. Neurobiol. Aging 1995, 16, 433–445. [Google Scholar] [CrossRef]
- Duyckaerts, C. Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol. 2011, 121, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Fasulo, L.; Visintin, M.; Novak, M.; Cattaneo, A. Tau truncation in Alzheimer’s disease: Expression of a fragment encompassing PHF core tau induces apoptosis in COS cells. Alzheimer’s Rep. 1998, 1, 25–32. [Google Scholar]
- Fasulo, L.; Ovecka, M.; Kabát, J.; Bradbury, A.; Novák, M.; Cattaneo, A. Overexpression of Alzheimer’s PHF core tau fragments: Implications for the tau truncation hypothesis. Alzheimer’s Res. 1996, 2, 195–200. [Google Scholar]
- Schweers, O.; Mandelkow, E.-M.; Biernat, J.; Mandelkow, E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein t controls the in vitro assembly of paired helical filaments. PNAS 1995, 92, 8463–8467. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. PNAS 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.Y.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Cavallini, A.; Angers, R.; Glover, S.; Murray, T.K.; Barnham, L.; Jackson, S.; O’Neill, M.J.; Isaacs, A.M.; Hutton, M.L.; et al. Conformation determines the seeding potencies of native and recombinant tau aggregates. J. Biol. Chem. 2015, 290, 1049–1065. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.T.; Sigurdsson, E.M. Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 2015, 21, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tau Protein | Abbreviation | Description |
|---|---|---|
| Truncated PHF-core tau | dGA | Bacterial recombinant human tau, residues 297–390 of 2N4R tau isoform [35]. |
| Full-length, non-phosphorylated tau | T40 | Bacterial recombinant human tau, 441-amino acid 2N4R tau isoform [33]. |
| Hyperphosphorylated tau | T40P | T40 tau hyperphosphorylated in vitro [36]. |
| Neonatal rat tau | NT | Endogenously phosphorylated neonatal rat 3-repeat tau. |
| Hyperphosphorylated rat tau | NTP | Rat NT hyperphosphorylated in vitro [36]. |
| Solid-Phase Species | Aqueous-Phase Species | Solid-Phase Species Non-Limiting | Aqueous-Phase Species Non-Limiting | ||||
| N1 | Q2 | Kd,Aqeous | N2 | Q1 | Kd,Adsorbed | ||
| dGA | T40 | 22.8 ± 2.8 | 19.3 ± 0.8 | 21.1 | 36.3 ± 21.4 | 26.6 ± 7.3 | 31.5 |
| dGA | T40P | 252.4 ± 69.8 | 220.5 ± 66.4 | 236.5 | 22.5 ± 17.7 | 34.9 ± 8.2 | 28.7 |
| dGA | NT | -* | - | - | - | - | - |
| NT | T40 | 18.8 ± 3.4 | 22.8 ± 9.9 | 20.8 | 8.6 ± 5.0 | 7.6 ± 1.5 | 8.1 |
| NT | T40P | 627 ± 131.0 | 382.1 ± 64.2 | 504.5 | 39.1 ± 4.9 | 23.8 ± 1.4 | 31.5 |
| NTP | T40 | 754 ± 181.0 | 758.8 ± 138.3 | 756.4 | 2.2 ± 1.3 | 18.8 ± 3.7 | 10.5 |
| NTP | T40P | 969.2 ± 8.8 | 1163 ± 223 | 1066.1 | 86 ± 8.0 | 114.7 ± 15.6 | 100.4 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, R.Y.K.; Harrington, C.R.; Wischik, C.M. Absence of a Role for Phosphorylation in the Tau Pathology of Alzheimer’s Disease. Biomolecules 2016, 6, 19. https://doi.org/10.3390/biom6020019
Lai RYK, Harrington CR, Wischik CM. Absence of a Role for Phosphorylation in the Tau Pathology of Alzheimer’s Disease. Biomolecules. 2016; 6(2):19. https://doi.org/10.3390/biom6020019
Chicago/Turabian StyleLai, Robert Y. K., Charles R. Harrington, and Claude M. Wischik. 2016. "Absence of a Role for Phosphorylation in the Tau Pathology of Alzheimer’s Disease" Biomolecules 6, no. 2: 19. https://doi.org/10.3390/biom6020019
APA StyleLai, R. Y. K., Harrington, C. R., & Wischik, C. M. (2016). Absence of a Role for Phosphorylation in the Tau Pathology of Alzheimer’s Disease. Biomolecules, 6(2), 19. https://doi.org/10.3390/biom6020019