
Oxidative Stress in Aging Human Skin

 ,
, {kind=link}
Abstract
:1. Introduction: The Skin as a Model for the “ROS-Aging” Connection
2. The Aging Process in the Dermis
3. The Epidermis: The Process of Cornification and Aging
3.1. The Cornified Envelope Formation
3.2. The Calcium Dependence of the Cornified Envelope Formation
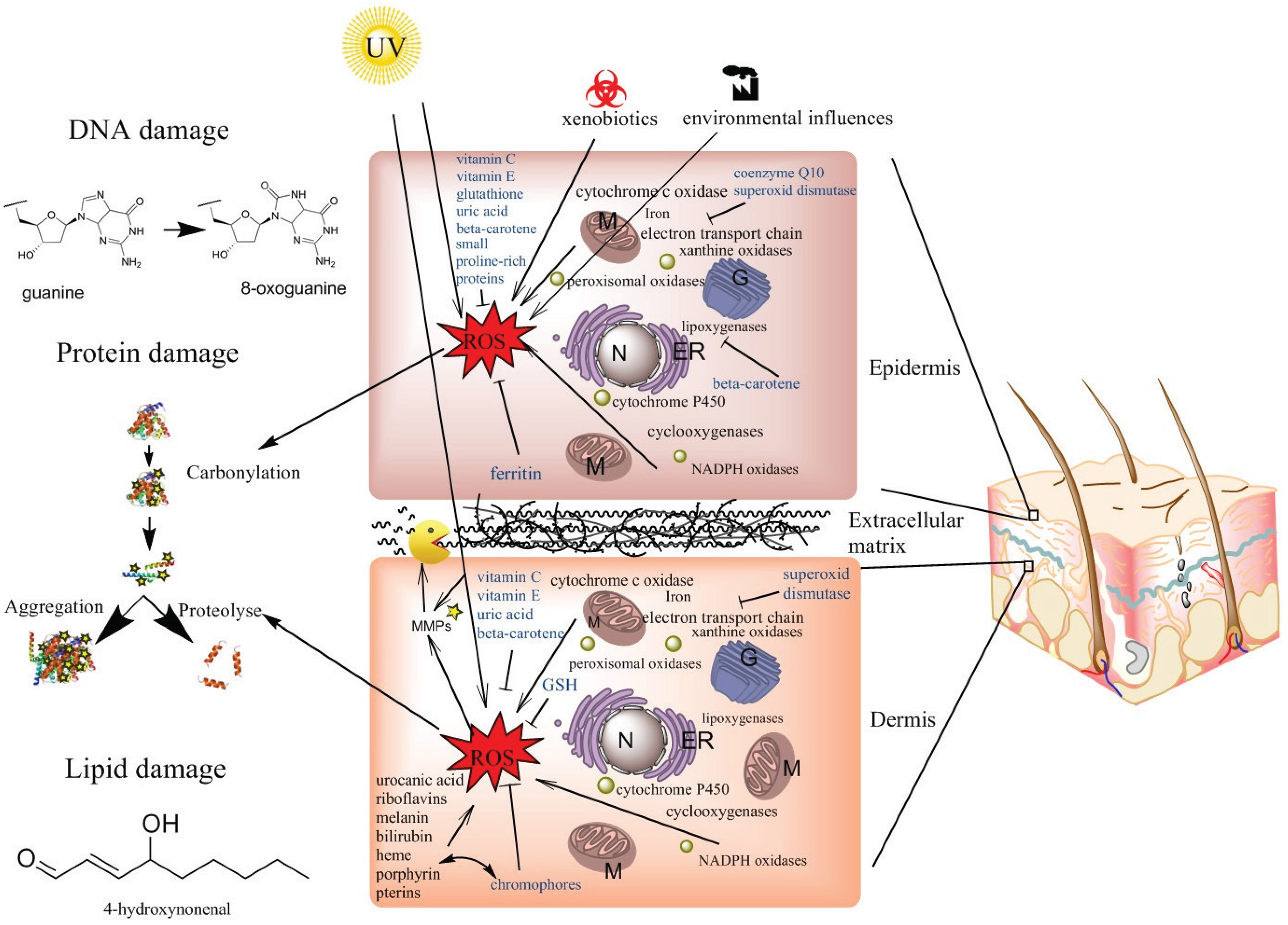
4. ROS Production in the Skin
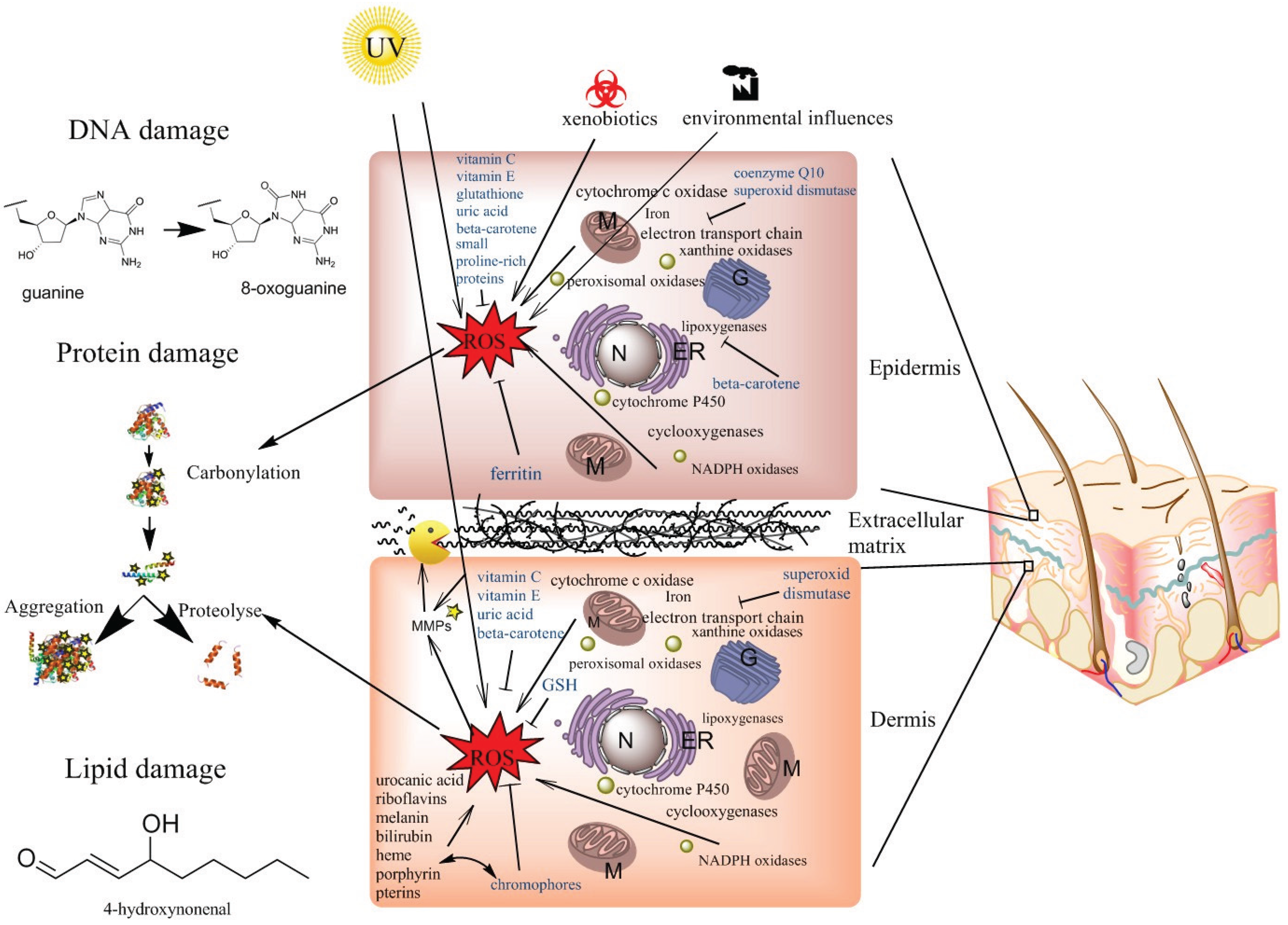
4.1. Mitochondrial ROS Production
4.2. Peroxisomal ROS Production
4.3. ROS Production in the Endoplasmic Reticulum
4.4. ROS Production in Membranes and in the Cytosol
4.5. Photoaging or UV-Induced ROS
5. Anti-Oxidative Capacities of the Skin
5.1. Anti-Oxidative Properties of the Cornified Envelope
5.2. The Non-Enzymatic Anti-Oxidants Vitamin C, Vitamin E, Beta-Carotene and CoQ10
5.3. The Importance of Superoxide Dismutases, Catalases, Glutathione Peroxidases, Ferritin, and Peroxiredoxins in Quenching ROS
5.4. The Anti-Oxidant Treatment Paradox
6. Protein Oxidation, Lipofuscin and AGEs
6.1. Protein Carbonyls
6.2. The Process of Protein Oxidation
6.3. Lipofuscin
6.4. Advanced Glycation End Products
7. Oxidative Stress, DNA, Cancer and Senescence
7.1. DNA Mutation
7.2. Cancer
7.3. Senescence
8. Oxidative Stress and Lipids
9. Conclusions
- the mitochondrial electron transport chain
- peroxisomal localized enzymes involved in the β-oxidation of fatty acids, the glyoxylate/dicarboxylate metabolism and the xanthine oxidase, involved in purine catabolism
- the endoplasmic reticulum, localized enzymes, protein disulfide isomerase, and endoplasmic reticulum oxidoreductin-1 ERO1 as well as members of the cytochrome P450 family
- the enzymes cyclooxygenase and lipoxygenase involved in the arachidonic acid metabolism, the Fenton reaction and the membrane localized NADPH oxidase family
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harman, D. Aging—A theory based on free-radical and radiation-chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [PubMed]
- Lapointe, J.; Hekimi, S. When a theory of aging ages badly. Cell. Mol. Life Sci. 2010, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S. How genetic analysis tests theories of animal aging. Nat. Genet. 2006, 38, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Poljsak, B.; Dahmane, R.G.; Godic, A. Intrinsic skin aging: The role of oxidative stress. Acta Dermatovenerol. Alp. Pannonica Adriat. 2012, 21, 33–36. [Google Scholar] [PubMed]
- Tsutsumi, M.; Denda, M. Paradoxical effects of beta-estradiol on epidermal permeability barrier homeostasis. Br. J. Dermatol. 2007, 157, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Chen, W.C.; Thornton, M.J.; Qin, K.; Rosenfield, R. Sexual hormones in human skin. Horm. Metab. Res. 2007, 39, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Makrantonaki, E. Clinical aspects and molecular diagnostics of skin aging. Clin. Dermatol. 2011, 29, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the aging skin. Adv. Wound care 2013, 2, 5–10. [Google Scholar] [CrossRef]
- Vierkotter, A.; Krutmann, J. Environmental influences on skin aging and ethnic-specific manifestations. Dermato Endocrinol. 2012, 4, 227–231. [Google Scholar] [CrossRef]
- Ritz-Timme, S.; Laumeier, I.; Collins, M.J. Aspartic acid racemization: Evidence for marked longevity of elastin in human skin. Br. J. Dermatol. 2003, 149, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of d-aspartate and nuclear weapons-related radiocarbon. J. Clin. Invest. 1991, 87, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Kielty, C.M.; Sherratt, M.J.; Shuttleworth, C.A. Elastic fibers. J. Cell Sci. 2002, 115, 2817–2828. [Google Scholar] [PubMed]
- Naylor, E.C.; Watson, R.E.; Sherratt, M.J. Molecular aspects of skin ageing. Maturitas 2011, 69, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Graham, H.K.; Hodson, N.W.; Hoyland, J.A.; Millward-Sadler, S.J.; Garrod, D.; Scothern, A.; Griffiths, C.E.; Watson, R.E.; Cox, T.R.; Erler, J.T.; et al. Tissue section AFM: In situ ultrastructural imaging of native biomolecules. Matrix Biol. 2010, 29, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Keene, D.R.; Sakai, L.Y.; Lunstrum, G.P.; Morris, N.P.; Burgeson, R.E. Type VII collagen forms an extended network of anchoring fibrils. J. Cell Biol. 1987, 104, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Craven, N.M.; Watson, R.E.; Jones, C.J.; Shuttleworth, C.A.; Kielty, C.M.; Griffiths, C.E. Clinical features of photodamaged human skin are associated with a reduction in collagen VII. Br. J. Dermatol. 1997, 137, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Talwar, H.S.; Griffiths, C.E.; Fisher, G.J.; Hamilton, T.A.; Voorhees, J.J. Reduced type I and type III procollagens in photodamaged adult human skin. J. Investig. Dermatol. 1995, 105, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Labat-Robert, J.; Fourtanier, A.; Boyer-Lafargue, B.; Robert, L. Age dependent increase of elastase type protease activity in mouse skin. Effect of UV-irradiation. J. Photochem. Photobiol. B Biol. 2000, 57, 113–118. [Google Scholar] [CrossRef]
- Birkedal-Hansen, H.; Moore, W.G.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: A review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [PubMed]
- Berton, A.; Godeau, G.; Emonard, H.; Baba, K.; Bellon, P.; Hornebeck, W.; Bellon, G. Analysis of the ex vivo specificity of human gelatinases a and b towards skin collagen and elastic fibers by computerized morphometry. Matrix Biol. 2000, 19, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. Molecular mechanisms of skin ageing. Mech. Ageing Dev. 2002, 123, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Millis, A.J.; Hoyle, M.; McCue, H.M.; Martini, H. Differential expression of metalloproteinase and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp. Cell Res. 1992, 201, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kohl, E.; Steinbauer, J.; Landthaler, M.; Szeimies, R.M. Skin ageing. J. Eur. Acad. Dermatol. 2011, 25, 873–884. [Google Scholar] [CrossRef]
- Lavker, R.M.; Veres, D.A.; Irwin, C.J.; Kaidbey, K.H. Quantitative assessment of cumulative damage from repetitive exposures to suberythemogenic doses of uva in human skin. Photochem. Photobiol. 1995, 62, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Smad7 via induction of transcription factor AP-1 in human skin fibroblasts. J. Biol. Chem. 2005, 280, 8079–8085. [Google Scholar] [CrossRef] [PubMed]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol. 2006, 126, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, T.; Campisi, J. Repression of c-Fos transcription and an altered genetic program in senescent human fibroblasts. Science 1990, 247, 205–209. [Google Scholar] [CrossRef]
- Campisi, J. Aging and cancer: The double-edged sword of replicative senescence. J. Am. Geriatr. Soc. 1997, 45, 482–488. [Google Scholar] [PubMed]
- Campisi, J. Cancer, aging and cellular senescence. In Vivo 2000, 14, 183–188. [Google Scholar] [PubMed]
- Tsatsou, F.; Trakatelli, M.; Patsatsi, A.; Kalokasidis, K.; Sotiriadis, D. Extrinsic aging: UV-mediated skin carcinogenesis. Dermato Endocrinol. 2012, 4, 285–297. [Google Scholar] [CrossRef]
- Samarasinghe, V.; Madan, V. Nonmelanoma skin cancer. J. Cutan. Aesthet. Surg. 2012, 5, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.R.; Pai, V.V. Novel medical strategies combating nonmelanoma skin cancer. Indian J. Dermatol. 2014, 59, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.H. Photocarcinogenesis, skin cancer, and aging. J. Am. Acad. Dermatol. 1983, 9, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Travers, J.B.; Somani, A.K.; Spandau, D.F. The IGF-1/IGF-1R signaling axis in the skin: A new role for the dermis in aging-associated skin cancer. Oncogene 2010, 29, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.L.; Grichnik, J.M.; Prieto, V.G.; Shea, C.R. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J. Cutan. Pathol. 1998, 25, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Quevedo, W.C.; Szabo, G.; Virks, J. Influence of age and UV on the populations of dopa-positive melanocytes in human skin. J. Investig. Dermatol. 1969, 52, 287–290. [Google Scholar] [PubMed]
- Sarin, K.Y.; Artandi, S.E. Aging, graying and loss of melanocyte stem cells. Stem Cell Rev. 2007, 3, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Passi, S.; Grandinetti, M.; Maggio, F.; Stancato, A.; de Luca, C. Epidermal oxidative stress in vitiligo. Pigment Cell Res. 1998, 11, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Ruhrberg, C.; Hajibagheri, M.A.; Simon, M.; Dooley, T.P.; Watt, F.M. Envoplakin, a novel precursor of the cornified envelope that has homology to desmoplakin. J. Cell Biol. 1996, 134, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Ruhrberg, C.; Hajibagheri, M.A.; Parry, D.A.; Watt, F.M. Periplakin, a novel component of cornified envelopes and desmosomes that belongs to the plakin family and forms complexes with envoplakin. J. Cell Biol. 1997, 139, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Green, H. Involucrin synthesis is correlated with cell size in human epidermal cultures. J. Cell Biol. 1981, 90, 738–742. [Google Scholar] [CrossRef] [PubMed]
- DiColandrea, T.; Karashima, T.; Maatta, A.; Watt, F.M. Subcellular distribution of envoplakin and periplakin: Insights into their role as precursors of the epidermal cornified envelope. J. Cell Biol. 2000, 151, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Nachat, R.; Groot, K.R.; Klement, J.F.; Uitto, J.; Djian, P.; Maatta, A.; Watt, F.M. Mice deficient in involucrin, envoplakin, and periplakin have a defective epidermal barrier. J. Cell Biol. 2007, 179, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Grayson, S.; Johnson-Winegar, A.G.; Wintroub, B.U.; Isseroff, R.R.; Epstein, E.H., Jr.; Elias, P.M. Lamellar body-enriched fractions from neonatal mice: Preparative techniques and partial characterization. J. Investig. Dermatol. 1985, 85, 289–294. [Google Scholar] [CrossRef]
- Chapman, S.J.; Walsh, A. Membrane-coating granules are acidic organelles which possess proton pumps. J. Investig. Dermatol. 1989, 93, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.A.; Gonzalez de Peredo, A.; Stella, A.; Ishida-Yamamoto, A.; Bouyssie, D.; Serre, G.; Monsarrat, B.; Simon, M. Lamellar bodies of human epidermis: Proteomics characterization by high throughput mass spectrometry and possible involvement of Clip-170 in their trafficking/secretion. Mol. Cell. Proteomics 2008, 7, 2151–2175. [Google Scholar] [CrossRef] [PubMed]
- Braff, M.H.; di Nardo, A.; Gallo, R.L. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J. Investig. Dermatol. 2005, 124, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Galliano, M.F.; Toulza, E.; Gallinaro, H.; Jonca, N.; Ishida-Yamamoto, A.; Serre, G.; Guerrin, M. A novel protease inhibitor of the alpha2-macroglobulin family expressed in the human epidermis. J. Biol. Chem. 2006, 281, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Madison, K.C.; Sando, G.N.; Howard, E.J.; True, C.A.; Gilbert, D.; Swartzendruber, D.C.; Wertz, P.W. Lamellar granule biogenesis: A role for ceramide glucosyltransferase, lysosomal enzyme transport, and the golgi. J. Investig. Dermatol. Sympos. Proc. 1998, 3, 80–86. [Google Scholar] [CrossRef]
- Serre, G.; Mils, V.; Haftek, M.; Vincent, C.; Croute, F.; Reano, A.; Ouhayoun, J.P.; Bettinger, S.; Soleilhavoup, J.P. Identification of late differentiation antigens of human cornified epithelia, expressed in re-organized desmosomes and bound to cross-linked envelope. J. Investig. Dermatol. 1991, 97, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Denda, M.; Fuziwara, S.; Inoue, K. Influx of calcium and chloride ions into epidermal keratinocytes regulates exocytosis of epidermal lamellar bodies and skin permeability barrier homeostasis. J. Investig. Dermatol. 2003, 121, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, A.; Marekov, L.N.; Steinert, P.M. Assembly of the epidermal cornified cell envelope. J. Cell Sci. 2001, 114, 3069–3070. [Google Scholar] [PubMed]
- Kalinin, A.E.; Idler, W.W.; Marekov, L.N.; McPhie, P.; Bowers, B.; Steinert, P.M.; Steven, A.C. Co-assembly of envoplakin and periplakin into oligomers and Ca2+-dependent vesicle binding: Implications for cornified cell envelope formation in stratified squamous epithelia. J. Biol. Chem. 2004, 279, 22773–22780. [Google Scholar] [CrossRef] [PubMed]
- Marekov, L.N.; Steinert, P.M. Ceramides are bound to structural proteins of the human foreskin epidermal cornified cell envelope. J. Biol. Chem 1998, 273, 17763–17770. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Eady, R.A.; Watt, F.M.; Roop, D.R.; Hohl, D.; Iizuka, H. Immunoelectron microscopic analysis of cornified cell envelope formation in normal and psoriatic epidermis. J. Histochem. Cytochem 1996, 44, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ahvazi, B.; Boeshans, K.M.; Idler, W.; Baxa, U.; Steinert, P.M. Roles of calcium ions in the activation and activity of the transglutaminase 3 enzyme. J. Biol. Chem. 2003, 278, 23834–23841. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Sturniolo, M.T.; Broome, A.M.; Ruse, M.; Rorke, E.A. Transglutaminase function in epidermis. J. Investig. Dermatol. 2005, 124, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Marekov, L.N. Initiation of assembly of the cell envelope barrier structure of stratified squamous epithelia. Mol. Biol. Cell 1999, 10, 4247–4261. [Google Scholar] [CrossRef] [PubMed]
- Hohl, D.; Lichti, U.; Breitkreutz, D.; Steinert, P.M.; Roop, D.R. Transcription of the human loricrin gene in vitro is induced by calcium and cell density and suppressed by retinoic acid. J. Investig. Dermatol. 1991, 96, 414–418. [Google Scholar] [CrossRef]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef]
- Whitfield, J.M. Calcium in Cell Cycles and Cancer, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1995; p. 242. [Google Scholar]
- Robinson, N.A.; Lapic, S.; Welter, J.F.; Eckert, R.L. S100a11, s100a10, annexin I, desmosomal proteins, small proline-rich proteins, plasminogen activator inhibitor-2, and involucrin are components of the cornified envelope of cultured human epidermal keratinocytes. J. Biol. Chem. 1997, 272, 12035–12046. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.; Hardman, M.J.; Nield, K.M.; Byrne, C. Differentially expressed late constituents of the epidermal cornified envelope. Proc. Natl. Acad. Sci. USA 2001, 98, 13031–13036. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Tilli, C.M.; Hardman, M.J.; Avilion, A.A.; MacLeod, M.C.; Ashcroft, G.S.; Byrne, C. Late cornified envelope family in differentiating epithelia—Response to calcium and ultraviolet irradiation. J. Investig. Dermatol. 2005, 124, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Declercq, W.; Ban, J.; Rendl, M.; Lengauer, B.; Mayer, C.; Lippens, S.; Vandenabeele, P.; Tschachler, E. Terminal differentiation of human keratinocytes and stratum corneum formation is associated with caspase-14 activation. J. Investig. Dermatol. 2000, 115, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Mauro, T.; Bench, G.; Sidderas-Haddad, E.; Feingold, K.; Elias, P.; Cullander, C. Acute barrier perturbation abolishes the Ca2+ and K+ gradients in murine epidermis: Quantitative measurement using pixe. J. Investig. Dermatol. 1998, 111, 1198–1201. [Google Scholar] [CrossRef]
- Menon, G.K.; Elias, P.M.; Lee, S.H.; Feingold, K.R. Localization of calcium in murine epidermis following disruption and repair of the permeability barrier. Cell Tissue Res. 1992, 270, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Duschl, J.; Steinbacher, P.; Salzmann, M.; Bischof, J.; Schuller, M.; Wimmer, H.; Peer, T.; Bauer, J.W.; Richter, K. Age-related changes in the composition of the cornified envelope in human skin. Exp. Dermatol. 2013, 22, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Streubel, M.K.; Bischof, J.; Richter, K. Skin aging, gene expression and calcium. Exp. Gerontol. 2014. [Google Scholar] [CrossRef]
- Denda, M.; Tomitaka, A.; Akamatsu, H.; Matsunaga, K. Altered distribution of calcium in facial epidermis of aged adults. J. Investig. Dermatol. 2003, 121, 1557–1558. [Google Scholar] [CrossRef] [PubMed]
- Lener, T.; Moll, P.R.; Rinnerthaler, M.; Bauer, J.; Aberger, F.; Richter, K. Expression profiling of aging in the human skin. Exp. Gerontol. 2006, 41, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Muck, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell 2010, 9, 291–296. [Google Scholar] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, K.A. Biologic structure and function—Perspectives on morphologic approaches to the study of the granular layer keratinocyte. J. Investig. Dermatol. 1989, 92, S84–S104. [Google Scholar]
- Jeon, S.; Djian, P.; Green, H. Inability of keratinocytes lacking their specific transglutaminase to form cross-linked envelopes: Absence of envelopes as a simple diagnostic test for lamellar ichthyosis. Proc. Natl. Acad. Sci. USA 1998, 95, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Breitenbach, M.; Rinnerthaler, M.; Hartl, J.; Stincone, A.; Vowinckel, J.; Breitenbach-Koller, H.; Ralser, M. Mitochondria in ageing: There is metabolism beyond the ROS. Fems Yeast Res. 2014, 14, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; Lam, Y.T.; Lim, Y.L.; Rinnerthaler, M.; Gelling, C.L.; Yang, H.Y.; Breitenbach, M.; Dawes, I.W. Maintenance of mitochondrial morphology by autophagy and its role in high glucose effects on chronological lifespan of saccharomyces cerevisiae. Oxid. Med. Cell Longev. 2013. [Google Scholar] [CrossRef]
- Klinger, H.; Rinnerthaler, M.; Lam, Y.T.; Laun, P.; Heeren, G.; Klocker, A.; Simon-Nobbe, B.; Dickinson, J.R.; Dawes, I.W.; Breitenbach, M. Quantitation of (a)symmetric inheritance of functional and of oxidatively damaged mitochondrial aconitase in the cell division of old yeast mother cells. Exp. Gerontol. 2010, 45, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- Kuppusamy, P.; Zweier, J.L. Characterization of free radical generation by xanthine oxidase. Evidence for hydroxyl radical generation. J. Biol. Chem 1989, 264, 9880–9884. [Google Scholar] [PubMed]
- Granger, D.N.; Hollwarth, M.E.; Parks, D.A. Ischemia-reperfusion injury: Role of oxygen-derived free radicals. Acta Physiol. Scand. Suppl. 1986, 548, 47–63. [Google Scholar] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Benham, A.M.; van Lith, M.; Sitia, R.; Braakman, I. Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Do Yoo, Y. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Gorsky, L.D.; Koop, D.R.; Coon, M.J. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J. Biol. Chem. 1984, 259, 6812–6817. [Google Scholar] [PubMed]
- Yengi, L.G.; Xiang, Q.; Pan, J.M.; Scatina, J.; Kao, J.; Ball, S.E.; Fruncillo, R.; Ferron, G.; Wolf, C.R. Quantitation of cytochrome p450 mrna levels in human skin. Analytical Biochem. 2003, 316, 103–110. [Google Scholar] [CrossRef]
- Baron, J.M.; Holler, D.; Schiffer, R.; Frankenberg, S.; Neis, M.; Merk, H.F.; Jugert, F.K. Expression of multiple cytochrome P450 enzymes and multidrug resistance-associated transport proteins in human skin keratinocytes. J. Investig. Dermatol. 2001, 116, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Buttner, S.; Laun, P.; Heeren, G.; Felder, T.K.; Klinger, H.; Weinberger, M.; Stolze, K.; Grousl, T.; Hasek, J.; et al. Yno1p/Aim14p, a NADPH-oxidase ortholog, controls extramitochondrial reactive oxygen species generation, apoptosis, and actin cable formation in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 8658–8663. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.H. Tissue distribution and putative physiological function of Nox family NADPH oxidases. Jpn. J. Infect Dis. 2004, 57, S28–S29. [Google Scholar] [PubMed]
- Nauseef, W.M. Biological roles for the Nox family NADPH oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Kontos, H.A.; Hess, M.L.; Ellis, E.F. Superoxide generation by prostaglandin synthetase and lipoxygenase in presence of NADH or NADPH. Fed. Proc. 1986, 45, 451–451. [Google Scholar]
- Ruzicka, T.; Simmet, T.; Peskar, B.A.; Ring, J. Skin levels of arachidonic acid-derived inflammatory mediators and histamine in atopic-dermatitis and psoriasis. J. Investig. Dermatol. 1986, 86, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ziboh, V.A.; Miller, C.C.; Cho, Y.H. Metabolism of polyunsaturated fatty acids by skin epidermal enzymes: Generation of antiinflammatory and antiproliferative metabolites. Am. J. Clin. Nutr. 2000, 71, 361s–366s. [Google Scholar] [PubMed]
- Barb, W.G.; Baxendale, J.H.; George, P.; Hargrave, K.R. Reactions of ferrous and ferric ions with hydrogen peroxide. Nature 1949, 163, 692–694. [Google Scholar] [CrossRef]
- Barbusinski, K. Fenton reaction—Controversy concerning the chemistry. Ecol. Chem. Eng. 2009, 16, 347–358. [Google Scholar]
- Poljsak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012. [Google Scholar] [CrossRef]
- Chen, L.; Hu, J.Y.; Wang, S.Q. The role of antioxidants in photoprotection: A critical review. J. Am. Acad. Dermatol. 2012, 67, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Pospisil, P. Ultraweak photon emission induced by visible light and ultraviolet a radiation via photoactivated skin chromophores: In vivo charge coupled device imaging. J. Biomed. Opt. 2012. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci. 2006, 5, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Berneburg, M.; Gattermann, N.; Stege, H.; Grewe, M.; Vogelsang, K.; Ruzicka, T.; Krutmann, J. Chronically ultraviolet-exposed human skin shows a higher mutation frequency of mitochondrial DNA as compared to unexposed skin and the hematopoietic system. J. Investig. Dermatol. 1997, 109, 425–425. [Google Scholar]
- Siddens, L.K.; Larkin, A.; Krueger, S.K.; Bradfield, C.A.; Waters, K.M.; Tilton, S.C.; Pereira, C.B.; Lohr, C.V.; Arlt, V.M.; Phillips, D.H.; et al. Polycyclic aromatic hydrocarbons as skin carcinogens: Comparison of benzo [a]pyrene, dibenzo[def,p]chrysene and three environmental mixtures in the FVB/N mouse. Toxicol. Appl. Pharmacol. 2012, 264, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.P.; Xia, Q.S.; Sun, X.; Yu, H.T. Phototoxicity and environmental transformation of polycyclic aromatic hydrocarbons (PAHs)-light-induced reactive oxygen species, lipid peroxidation, and DNA damage. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2012, 30, 1–41. [Google Scholar] [CrossRef]
- Vermeij, W.P.; Alia, A.; Backendorf, C. ROS quenching potential of the epidermal cornified cell envelope. J. Investig. Dermatol. 2011, 131, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.U.; Thiele, J.J.; Cross, C.E.; Packer, L. Vitamin C, uric acid, and glutathione gradients in murine stratum corneum and their susceptibility to ozone exposure. J. Investig. Dermatol. 1999, 113, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. The cornified envelope: A first line of defense against reactive oxygen species. J. Investig. Dermatol. 2011, 131, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Dutsch, S.; Keller, U.A.D.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of uvb cytoprotection in the epidermis. Gene Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Traber, M.G.; Packer, L. Depletion of human stratum corneum vitamin E: An early and sensitive in vivo marker of UV induced photo-oxidation. J. Investig. Dermatol. 1998, 110, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Colven, R.M.; Pinnell, S.R. Topical vitamin C in aging. Clin. Dermatol. 1996, 14, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.H.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.L.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Humbert, P.G.; Haftek, M.; Creidi, P.; Lapiere, C.; Nusgens, B.; Richard, A.; Schmitt, D.; Rougier, A.; Zahouani, H. Topical ascorbic acid on photoaged skin. Clinical, topographical and ultrastructural evaluation: Double-blind study vs. Placebo. Exp. Dermatol. 2003, 12, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Rhie, G.; Shin, M.H.; Seo, J.Y.; Choi, W.W.; Cho, K.H.; Kim, K.H.; Park, K.C.; Eun, H.C.; Chung, J.H. Aging- and photoaging-dependent changes of enzymic and nonenzymic antioxidants in the epidermis and dermis of human skin in vivo. J. Investig. Dermatol. 2001, 117, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Nachbar, F.; Korting, H.C. The role of vitamin-E in normal and damaged skin. J. Mol. Med. 1995, 73, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H.; Radi, R.; Anselmi, D.; Kirk, M.; Barnes, S.; Butler, J.; Eiserich, J.P.; Freeman, B.A. Nitric oxide reaction with lipid peroxyl radicals spares alpha-tocopherol during lipid peroxidation—Greater oxidant protection from the pair nitric oxide/alpha-tocopherol than alpha-tocopherol/ascorbate. J. Biol. Chem. 2000, 275, 10812–10818. [Google Scholar] [CrossRef] [PubMed]
- Engin, K.N. Alpha-tocopherol: Looking beyond an antioxidant. Mol. Vis. 2009, 15, 855–860. [Google Scholar] [PubMed]
- Bayerl, C. Beta-carotene in dermatology: Does it help? Acta Dermatovenerol. Alp. Pannon. Adriat. 2008, 17, 160–162. [Google Scholar]
- BarNatan, R.; Lomnitski, L.; Sofer, Y.; Segman, S.; Neeman, I.; Grossman, S. Interaction between beta-carotene and lipoxygenase in human skin. Int. J. Biochem. Cell Biol. 1996, 28, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Liebler, D.C.; McClure, T.D. Antioxidant reactions of beta-carotene: Identification of carotenoid-radical adducts. Chem. Res. Toxicol. 1996, 9, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Maples, K.R.; Mason, R.P. Free-radical metabolite of uric-acid. J. Biol. Chem. 1988, 263, 1709–1712. [Google Scholar] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric-acid provides an antioxidant defense in humans against oxidant-caused and radical-caused aging and cancer—A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Tzagoloff, A. The mitochondrial electron transfer chain. Arch. Biochem. Biophys. 1966, 116, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Littarru, G.P.; Tiano, L. Bioenergetic and antioxidant properties of coenzyme Q10: Recent developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Kwon, M.J.; Kim, B.; Lee, Y.S.; Kim, T.Y. Role of superoxide dismutase 3 in skin inflammation. J. Dermatol. Sci. 2012, 67, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Rulisek, L.; Ryde, U. Structure of reduced and oxidized manganese superoxide dismutase: A combined computational and experimental approach. J. Phys. Chem. B 2006, 110, 11511–11518. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic sod-deficient mice and its complete recovery using a vitamin c derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, S.; Ozawa, Y.; Watanabe, K.; Izuo, N.; Toda, T.; Yokote, K.; Shimizu, T. Palladium and platinum nanoparticles attenuate aging-like skin atrophy via antioxidant activity in mice. PLOS ONE 2014, 9, e109288. [Google Scholar] [CrossRef] [PubMed]
- Poswig, A.; Wenk, J.; Brenneisen, P.; Wlaschek, M.; Hommel, C.; Quel, G.; Faisst, K.; Dissemond, J.; Briviba, K.; Krieg, T.; et al. Adaptive antioxidant response of manganese-superoxide dismutase following repetitive uva irradiation. J. Investig. Dermatol. 1999, 112, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Perez, V.I.; Song, W.; Lustgarten, M.S.; Salmon, A.B.; Mele, J.; Qi, W.; Liu, Y.; Liang, H.; Chaudhuri, A.; et al. Overexpression of MN superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [PubMed]
- Treiber, N.; Maity, P.; Singh, K.; Kohn, M.; Keist, A.F.; Ferchiu, F.; Sante, L.; Frese, S.; Bloch, W.; Kreppel, F.; et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011, 10, 912–912. [Google Scholar] [CrossRef]
- Treiber, N.; Maity, P.; Singh, K.; Ferchiu, F.; Wlaschek, M.; Scharffetter-Kochanek, K. The role of manganese superoxide dismutase in skin aging. Dermato Endocrinol. 2012, 4, 232–235. [Google Scholar] [CrossRef]
- Wagener, F.A.; Carels, C.E.; Lundvig, D.M. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [CrossRef] [PubMed]
- Loew, O. A new enzyme of general occurrence in organismis. Science 1900, 11, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Fita, I.; Silva, A.M.; Murthy, M.R.N.; Rossmann, M.G. The refined structure of beef-liver catalase at 2.5 Å resolution. Acta Crystallogr. Sect. B Struct. Sci. 1986, 42, 497–515. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnes, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, P.U.; Declercq, L.; Hellemans, L.; Maes, D. Aging of human skin: Review of a mechanistic model and first experimental data. IUBMB Life 2000, 49, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Rhie, G.E.; Kim, Y.K.; Park, C.H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes map kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; Ayer, A.; Breitenbach, M.; Dawes, I.W. Oxidative stresses and ageing. Subcell. Biochem. 2012, 57, 13–54. [Google Scholar] [PubMed]
- Lopez-Torres, M.; Shindo, Y.; Packer, L. Effect of age on antioxidants and molecular markers of oxidative damage in murine epidermis and dermis. J. Investig. Dermatol. 1994, 102, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L.; Toppo, S.; Cozza, G.; Ursini, F. A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Rozovsky, S. Glutathione peroxidases reaction intermediate selenenic acid is stabilized by the protein microenvironment. Free Radic. Biol. Med. 2014, 76, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Lichti, U.F.; Carlson, B.A.; Cataisson, C.; Ryscavage, A.O.; Mikulec, C.; Conrad, M.; Fischer, S.M.; Hatfield, D.L.; Yuspa, S.H. Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of cox-2. J. Investig. Dermatol. 2013, 133, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Applegate, L.A.; Scaletta, C.; Panizzon, R.; Frenk, E. Evidence that ferritin is uv inducible in human skin: Part of a putative defense mechanism. J. Investig. Dermatol. 1998, 111, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Applegate, L.A.; Frenk, E. Oxidative defense in cultured human skin fibroblasts and keratinocytes from sun-exposed and non-exposed skin. Photodermatol. Photoimmunol. Photomed. 1995, 11, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Pourzand, C.; Watkin, R.D.; Brown, J.E.; Tyrrell, R.M. Ultraviolet a radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin. Proc. Natl. Acad. Sci. USA 1999, 96, 6751–6756. [Google Scholar] [CrossRef] [PubMed]
- Pelle, E.; Jian, J.; Zhang, Q.; Muizzuddin, N.; Yang, Q.; Dai, J.; Maes, D.; Pernodet, N.; Yarosh, D.B.; Frenkel, K.; et al. Menopause increases the iron storage protein ferritin in skin. J. Cosmet. Sci. 2013, 64, 175–179. [Google Scholar] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Chae, H.Z.; Lee, J.E.; Kwon, B.D.; Lee, J.B.; Won, Y.H.; Ahn, K.Y.; Kim, Y.P. Peroxiredoxin is ubiquitously expressed in rat skin: Isotype-specific expression in the epidermis and hair follicle. J. Investig. Dermatol. 2000, 115, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kimura, S.; Seto, K.; Warabi, E.; Kawachi, Y.; Shoda, J.; Tabuchi, K.; Yamagata, K.; Hasegawa, S.; Bukawa, H.; et al. Peroxiredoxin I plays a protective role against uva irradiation through reduction of oxidative stress. J. Dermatol. Sci. 2014, 74, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, F.; Huber, M.; Gruber, F.; Bohm, F.; Pfister, H.J.; Bochkov, V.N.; Tschachler, E.; Dummer, R.; Hohl, D.; Schafer, M.; et al. Dual role of the antioxidant enzyme peroxiredoxin 6 in skin carcinogenesis. Cancer Res. 2013, 73, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Kumin, A.; Huber, C.; Rulicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006, 169, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T.; Yang, J.S.; Molin, M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Gene Dev. 2012, 26, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Townley, J.P.; Barnes, T.M.; Greive, K.A. An antiaging skin care system containing alpha hydroxy acids and vitamins improves the biomechanical parameters of facial skin. Clin. Cosmet. Investig. Dermatol. 2015, 8, 9–17. [Google Scholar] [PubMed]
- Farris, P.; Yatskayer, M.; Chen, N.; Krol, Y.; Oresajo, C. Evaluation of efficacy and tolerance of a nighttime topical antioxidant containing resveratrol, baicalin, and vitamin e for treatment of mild to moderately photodamaged skin. J. Drugs Dermatol. 2014, 13, 1467–1472. [Google Scholar] [PubMed]
- Soeur, J.; Eilstein, J.; Lereaux, G.; Jones, C.; Marrot, L. Skin resistance to oxidative stress induced by resveratrol: From Nrf2 activation to gsh biosynthesis. Free Radic. Biol. Med. 2015, 78, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Lohan, S.B.; Bauersachs, S.; Ahlberg, S.; Baisaeng, N.; Keck, C.M.; Muller, R.H.; Witte, E.; Wolk, K.; Hackbarth, S.; Roder, B.; et al. Ultra-small lipid nanoparticles promote the penetration of coenzyme Q10 in skin cells and counteract oxidative stress. Eur. J. Pharm. Biopharm. 2015, 89, 201–207. [Google Scholar] [CrossRef]
- Schwarz, J.C.; Baisaeng, N.; Hoppel, M.; Low, M.; Keck, C.M.; Valenta, C. Ultra-small NLC for improved dermal delivery of coenyzme Q10. Int. J. Pharm. 2013, 447, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Lorencini, M.; Brohem, C.A.; Dieamant, G.C.; Zanchin, N.I.; Maibach, H.I. Active ingredients against human epidermal aging. Ageing Res. Rev. 2014, 15, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Bendall, J.K.; Scheiermann, C.; Watschinger, K.; Hoffmann, J.; Roeder, T.; Rimbach, G. Vitamin C and lifespan in model organisms. Food Chem. Toxicol. 2013, 58, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ernst, I.M.; Pallauf, K.; Bendall, J.K.; Paulsen, L.; Nikolai, S.; Huebbe, P.; Roeder, T.; Rimbach, G. Vitamin E supplementation and lifespan in model organisms. Ageing Res. Rev. 2013, 12, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of antioxidants supplementation on aging and longevity. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef]
- Selman, C.; McLaren, J.S.; Collins, A.R.; Duthie, G.G.; Speakman, J.R. Deleterious consequences of antioxidant supplementation on lifespan in a wild-derived mammal. Biol. Lett. 2013, 9. [Google Scholar] [CrossRef]
- Lam, Y.T.; Stocker, R.; Dawes, I.W. The lipophilic antioxidants alpha-tocopherol and coenzyme Q10 reduce the replicative lifespan of saccharomyces cerevisiae. Free Radic. Biol. Med. 2010, 49, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Shetty, R.A.; Forster, M.J.; Sumien, N. Coenzyme Q(10) supplementation reverses age-related impairments in spatial learning and lowers protein oxidation. Age 2013, 35, 1821–1834. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Senoo-Matsuda, N.; Miyake, K.; Yasuda, K.; Ishii, T.; Hartman, P.S.; Furukawa, S. Coenzyme Q10 can prolong C. Elegans lifespan by lowering oxidative stress. Mech. Ageing Dev. 2004, 125, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Marosz, A.; Chlubek, D. The risk of abuse of vitamin supplements. Ann. Acad. Medicae Stetin. 2014, 60, 60–64. [Google Scholar]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Heck, D.E.; Vetrano, A.M.; Mariano, T.M.; Laskin, J.D. UVB light stimulates production of reactive oxygen species—Unexpected role for catalase. J. Biol. Chem. 2003, 278, 22432–22436. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, J.B.; Pillai, E.D.; Jaeger, T.D.; Duncan, M.A. Ultraviolet and infrared photodissociation of si+(C6H6)(n) and si+(C6H6)(n)Ar clusters. J. Phys. Chem. A 2005, 109, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.S.; Chang, H.; Salzmann, S.; Muller, C.S.L.; Ekanayake-Mudiyanselage, S.; Elsner, P.; Thiele, J.J. Photoaging is associated with protein oxidation in human skin in vivo. J. Investig. Dermatol. 2002, 118, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Squier, T.C. Oxidative stress and protein aggregation during biological aging. Exp. Gerontol. 2001, 36, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.V.; Dean, R.T.; Wolff, S.P. Hydroxyl radical production and autoxidative glycosylation—Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes-mellitus and aging. Biochem. J. 1988, 256, 205–212. [Google Scholar] [PubMed]
- Wolff, S.P.; Wang, G.M.; Spector, A. Pro-oxidant activation of ocular reductants. 1. Copper and riboflavin stimulate ascorbate oxidation causing lens epithelial cytotoxicity in vitro. Exp. Eye Res. 1987, 45, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein oxidation and aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Holmgren, A. Glutaredoxins: Glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004, 6, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J. Methionine sulfoxide reductases: Ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim. Biophys. Acta Proteins Proteom 2005, 1703, 213–219. [Google Scholar] [CrossRef]
- Hohn, A.; Konig, J.; Grune, T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteomics 2013, 92, 132–159. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Traber, M.G.; Re, R.; Espuno, N.; Yan, L.J.; Cross, C.E.; Packer, L. Macromolecular carbonyls in human stratum corneum: A biomarker for environmental oxidant exposure? FEBS Lett. 1998, 422, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Avezov, K.; Reznick, A.Z.; Aizenbud, D. Time and dose effects of cigarette smoke and acrolein on protein carbonyl formation in hacat keratinocytes. Adv. Exp. Med. Biol. 2015, 849, 57–64. [Google Scholar] [PubMed]
- Thiele, J.J.; Schroeter, C.; Hsieh, S.N.; Podda, M.; Packer, L. The antioxidant network of the stratum corneum. Curr. Probl. Dermatol. 2001, 29, 26–42. [Google Scholar] [PubMed]
- Maisonneuve, E.; Ducret, A.; Khoueiry, P.; Lignon, S.; Longhi, S.; Talla, E.; Dukan, S. Rules governing selective protein carbonylation. PLOS ONE 2009, 4, e7269. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Marcocci, L.; Liu, L.L.; Suzuki, Y.J. Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 2010, 12, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Friguet, B. Changes of the proteasomal system during the aging process. Prog. Mol. Biol. Transl. Sci. 2012, 109, 249–275. [Google Scholar] [PubMed]
- Grune, T.; Merker, K.; Jung, T.; Sitte, N.; Davies, K.J.A. Protein oxidation and degradation during postmitotic senescence. Free Radic. Biol. Med. 2005, 39, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.A.; Woulfe, J. Lipofuscin and aging: A matter of toxic waste. Sci. Aging Knowl. Environ. 2005. [Google Scholar] [CrossRef]
- Jung, T.; Hohn, A.; Catalgol, B.; Grune, T. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch. Biochem. Biophys. 2009, 483, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Dreher, F.; Packer, L. Antioxidant defense systems in skin (reprinted from cosmeceuticals: Drugs vs. Cosmetics, pg 145–187, 2000). J. Toxicol. Cutaneous. Ocul. Toxicol. 2002, 21, 119–160. [Google Scholar] [CrossRef]
- Naskalski, J.W.; Bartosz, G. Oxidative modifications of protein structures. Adv. Clin. Chem. 2000, 35, 161–253. [Google Scholar] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J.A. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R.E.; Kono, Y.; Davies, K.J. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 1993, 268, 15405–15411. [Google Scholar] [PubMed]
- Buchberger, A.; Bukau, B.; Sommer, T. Protein quality control in the cytosol and the endoplasmic reticulum: Brothers in arms. Mol. Cell 2010, 40, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Dimayuga, E.; Keller, J.N. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid. Redox Signal. 2006, 8, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Hiller, M.M.; Finger, A.; Schweiger, M.; Wolf, D.H. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 1996, 273, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Catalgol, B.; Ziaja, I.; Breusing, N.; Jung, T.; Hohn, A.; Alpertunga, B.; Schroeder, P.; Chondrogianni, N.; Gonos, E.S.; Petropoulos, I.; et al. The proteasome is an integral part of solar ultraviolet a radiation-induced gene expression. J. Biol. Chem. 2009, 284, 30076–30086. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Shimizu, T.; Nishihira, J.; Abe, R.; Nakayama, T.; Taniguchi, M.; Sabe, H.; Ishibashi, T.; Shimizu, H. Ultraviolet a-induced production of matrix metalloproteinase-1 is mediated by macrophage migration inhibitory factor (MIF) in human dermal fibroblasts. J. Biol. Chem. 2004, 279, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, Y.; Lee, D.H.; Kim, Y.; Cho, K.H.; Chung, J.H. Basal and UV-induced MMP-1 expression are inhibited by p53 in human dermal fibroblasts. Exp. Dermatol. 2008, 17, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Reinke, L.A.; Kotake, Y.; Moore, D.R.; Nanji, A.A. Free radical formation during ketamine anesthesia in rats: A cautionary note. Free Radic. Biol. Med. 1998, 24, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. The important role of lipid peroxidation processes in aging and age dependent diseases. Mol. Biotechnol. 2007, 37, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.R.; Fu, M.X.; Ahmed, M.U.; Jenkins, A.J.; Lyons, T.J.; Thorpe, S.R. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol. Dial. Transplant. 1996, 11, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Sitte, N.; Huber, M.; Grune, T.; Ladhoff, A.; Doecke, W.D.; Von Zglinicki, T.; Davies, K.J.A. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. 2000, 14, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Carrard, G.; Bulteau, A.L.; Petropoulos, I.; Friguet, B. Impairment of proteasome structure and function in aging. Int. J. Biochem. Cell B 2002, 34, 1461–1474. [Google Scholar] [CrossRef]
- Hohn, A.; Jung, T.; Grimm, S.; Catalgol, B.; Weber, D.; Grune, T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic. Biol. Med. 2011, 50, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J.A. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and “aggresomes” during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Poppek, D.; Grune, T. protein oxidation and proteolysis during cellular senescence. Z. Gerontol. Geriatr. 2004, 37, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Koziel, R.; Greussing, R.; Maier, A.B.; Declercq, L.; Jansen-Durr, P. Functional interplay between mitochondrial and proteasome activity in skin aging. J. Investig. Dermatol. 2011, 131, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Robison, W.G. What is lipofuscin? Defining characteristics and differentiation from other autofluoreseent lysosomal storage bodies. Arch. Gerontol. Geriat 2002, 34, 169–184. [Google Scholar] [CrossRef]
- Soroka, Y.; Ma’or, Z.; Leshem, Y.; Verochovsky, L.; Neuman, R.; Bregegere, F.M.; Milner, Y. Aged keratinocyte phenotyping: Morphology, biochemical markers and effects of dead sea minerals. Exp. Gerontol. 2008, 43, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.; Jacobowitz, D.; Marks, B.H. A simple method for the demonstration of lipofuscin pigment. J. Histochem. Cytochem. 1960, 8, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Szweda, P.A.; Camouse, M.; Lundberg, K.C.; Oberley, T.D.; Szweda, L.I. Aging, lipofuscin formation, and free radical-mediated inhibition of cellular proteolytic systems. Ageing Res. Rev. 2003, 2, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Benavides, S.H.; Monserrat, A.J.; Farina, S.; Porta, E.A. Sequential histochemical studies of neuronal lipofuscin in human cerebral cortex from the first to the ninth decade of life. Arch. Gerontol. Geriatr. 2002, 34, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Jolly, S.S.; Brownstein, S.; Jordan, D.R.; Munro, S.M.; Keystone, E.C. Scleritis in a patient with limited wegener’s granulomatosis and takayasu’s arteritis. Can. J. Ophthalmol. 1995, 30, 371–373. [Google Scholar] [PubMed]
- Winterbourn, C.C.; Vile, G.F.; Monteiro, H.P. Ferritin, lipid peroxidation and redox-cycling xenobiotics. Free Radic. Res. Commun. 1991, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.Z. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic. Biol. Med. 1996, 21, 871–888. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Engels, M.; Klotz, L.O.; Kroncke, K.D.; Grune, T. Nitrotyrosine and protein carbonyls are equally distributed in HT22 cells after nitrosative stress. Free Radic. Biol. Med. 2007, 42, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Pompella, A.; Cambiaggi, C.; Dominici, S.; Paolicchi, A.; Tongiani, R.; Comporti, M. Single-cell investigation by laser scanning confocal microscopy of cytochemical alterations resulting from extracellular oxidant challenge. Histochem. Cell Biol. 1996, 105, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of intralysosomal ph in living cells and perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Sittig, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic. Biol. Med. 2012, 53, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging—When “all you can eat” is yourself. Sci. Aging Knowl. Environ. 2003. [Google Scholar] [CrossRef]
- Overbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chang, C.; Huang, R.; Liu, B.; Bao, L.; Liu, W. Ap1 is essential for generation of autophagosomes from the trans-golgi network. J. Cell Sci. 2012, 125, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy. Methods Mol. Biol. 2008, 445, 227–244. [Google Scholar] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Rodriguez-Colman, M.J.; Tamarit, J.; Ortega, Z.; Lucas, J.J.; Ferrer, I.; Ros, J.; Cabiscol, E. Protein oxidation in huntington disease affects energy production and vitamin B6 metabolism. Free Radic. Biol. Med. 2010, 49, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, J.; Gillespie, M.; Miller, R. Cutaneous amyloidosis. Skinmed 2011, 9, 96–100. [Google Scholar] [PubMed]
- Wang, Y.; Bruce, A.T.; Tu, C.; Ma, K.; Zeng, L.; Zheng, P.; Liu, Y. Protein aggregation of SERCA2 mutants associated with darier disease elicits ER stress and apoptosis in keratinocytes. J. Cell Sci. 2011, 124, 3568–3580. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.C. The action of amino acids on sugar; the formation of melanoidin by a methodic route. Cr. Hebd. Acad. Sci. 1912, 154, 66–68. [Google Scholar]
- Lo, T.W.C.; Westwood, M.E.; Mclellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions—A kinetic and mechanistic study with N-alpha-acetylarginine, N-alpha-acetylcysteine, and N-alpha-acetyllysine, and bovine serum-albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [PubMed]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.; Cai, W.J.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and age/rage-mediated cellular dysfunction affect the aging process—A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar]
- Beisswenger, P.J.; Howell, S.; Mackenzie, T.; Corstjens, H.; Muizzuddin, N.; Matsui, M.S. Two fluorescent wavelengths, 440ex/520em nm and 370ex/440em nm, reflect advanced glycation and oxidation end products in human skin without diabetes. Diabetes Technol. Ther. 2012, 14, 285–292. [Google Scholar] [CrossRef]
- Hyogo, H.; Yamagishi, S. Advanced glycation end products (ages) and their involvement in liver disease. Curr. Pharm. Des. 2008, 14, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bekker, P.; Tsimikas, S. Advanced glycation end products and diabetic cardiovascular disease. Cardiol. Rev. 2012, 20, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding rage, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Lohwasser, C.; Neureiter, D.; Weigle, B.; Kirchner, T.; Schuppan, D. The receptor for advanced glycation end products is highly expressed in the skin and upregulated by advanced glycation end products and tumor necrosis factor-alpha. J. Investig. Dermatol. 2006, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Bohm, M. Advanced glycation end products: Key players in skin aging? Dermato Endocrinol. 2012, 4, 259–270. [Google Scholar] [CrossRef]
- Sajithlal, G.B.; Chithra, P.; Chandrakasan, G. Advanced glycation end products induce crosslinking of collagen in vitro. Biochim. Biophys. Acta Mol. Basis Dis. 1998, 1407, 215–224. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshikawa, H.; Saruwatari, K.; Akazawa, Y.; Inoue, T.; Kuze, T.; Sayo, T.; Uchida, N.; Sugiyama, Y. The presence of N-epsilon-(carboxymethyl) lysine in the human epidermis. Biochim. Biophys. Acta Proteins Proteom 2011, 1814, 1246–1252. [Google Scholar] [CrossRef]
- Bucala, R.; Mitchell, R.; Arnold, K.; Innerarity, T.; Vlassara, H.; Cerami, A. Identification of the major site of apolipoprotein-B modification by advanced glycosylation end-products blocking uptake by the low-density-lipoprotein receptor. J. Biol. Chem. 1995, 270, 10828–10832. [Google Scholar] [CrossRef] [PubMed]
- Lal, M.A.; Brismar, H.; Eklof, A.C.; Aperia, A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002, 61, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (ages) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Loughlin, D.T.; Artlett, C.M. Precursor of advanced glycation end products mediates ER-stress-induced caspase-3 activation of human dermal fibroblasts through NAD(P)H oxidase 4. PLOS ONE 2010, 5, e11093. [Google Scholar] [CrossRef] [PubMed]
- Pageon, H.; Bakala, H.; Monnier, V.M.; Asselineau, D. Collagen glycation triggers the formation of aged skin in vitro. Eur. J. Dermatol. 2007, 17, 12–20. [Google Scholar] [PubMed]
- Zhu, P.; Yang, C.; Chen, L.H.; Ren, M.; Lao, G.J.; Yan, L. Impairment of human keratinocyte mobility and proliferation by advanced glycation end products-modified BSA. Arch. Dermatol. Res. 2011, 303, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Franke, S.; Sommer, M.; Henle, T.; Stein, G. Differences in the modulating potential of advanced glycation end product (age) peptides versus age proteins. Kidney Int. Suppl. 2001, 78, S63–S66. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and rage: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Chung, M.H.; Jones, D.S.; Inoue, H.; Ishikawa, H.; Kamiya, H.; Ohtsuka, E.; Nishimura, S. 8-hydroxyguanine, a DNA adduct formed by oxygen radicals: Its implication on oxygen radical-involved mutagenesis/carcinogenesis. J. Toxicol. Sci. 1991, 16, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sauvaigo, S.; Caillat, S.; Odin, F.; Nkengne, A.; Bertin, C.; Oddos, T. Effect of aging on DNA excision/synthesis repair capacities of human skin fibroblasts. J. Investig. Dermatol. 2010, 130, 1739–1741. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, M.; Sakumi, K.; Tominaga, Y.; Budiyanto, A.; Ueda, M.; Ichihashi, M.; Nakabeppu, Y.; Nishigori, C. 8-oxoguanine formation induced by chronic uvb exposure makes OGG1 knockout mice susceptible to skin carcinogenesis. Cancer Res. 2005, 65, 6006–6010. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.; Gan, W.; Shi, F.; Hu, G.X.; Chen, L.G.; Hayakawa, H.; Sekiguchi, M.; Cai, J.P. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxid. Med. Cell Longev. 2013. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Tewari, A.; Sarkany, R.P.; Young, A.R. Uva1 induces cyclobutane pyrimidine dimers but not 6–4 photoproducts in human skin in vivo. J. Investig. Dermatol. 2012, 132, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Jonason, A.; Simon, J.; Leffell, D.; Brash, D.E. Tumor suppressor gene mutations and photocarcinogenesis. Photochem. Photobiol. 1996, 63, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Otoshi, E.; Yagi, T.; Mori, T.; Matsunaga, T.; Nikaido, O.; Kim, S.T.; Hitomi, K.; Ikenaga, M.; Todo, T. Respective roles of cyclobutane pyrimidine dimers, (6–4)photoproducts, and minor photoproducts in ultraviolet mutagenesis of repair-deficient xeroderma pigmentosum a cells. Cancer Res. 2000, 60, 1729–1735. [Google Scholar] [PubMed]
- Todo, T.; Ryo, H.; Borden, A.; Lawrence, C.; Sakaguchi, K.; Hirata, H.; Nomura, T. Non-mutagenic repair of (6–4) photoproducts by (6–4) photolyase purified from drosophila melanogaster. Mutat. Res. DNA Repair. 1997, 385, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6–4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Tursun, M.; Duckett, D.R.; Drummond, J.T.; Modrich, P.; Sancar, A. Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol. Cell. Biol. 1997, 17, 760–769. [Google Scholar] [PubMed]
- Sancar, A. Structure and function of DNA photolyase. Biochemistry 1994, 33, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Pandel, R.; Poljsak, B.; Godic, A.; Dahmane, R. Skin photoaging and the role of antioxidants in its prevention. ISRN Dermatol. 2013. [Google Scholar] [CrossRef]
- Andressoo, J.O.; Mitchell, J.R.; de Wit, J.; Hoogstraten, D.; Volker, M.; Toussaint, W.; Speksnijder, E.; Beems, R.B.; van Steeg, H.; Jans, J.; et al. An xpd mouse model for the combined xeroderma pigmentosum/cockayne syndrome exhibiting both cancer predisposition and segmental progeria. Cancer Cell 2006, 10, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Bohr, V.A.; Sander, M.; Kraemer, K.H. Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: Molecules to patients. Mech. ageing Dev. 2011, 132, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Pouget, J.P.; Ravanat, J.L. Singlet oxygen DNA damage products: Formation and measurement. Methods Enzymol. 2000, 319, 143–153. [Google Scholar] [PubMed]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Goode, E.L.; Ladiges, W.C.; Ulrich, C.M. Polymorphic variation in hogg1 and risk of cancer: A review of the functional and epidemiologic literature. Mol. Carcinog. 2005, 42, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Diakowska, D.; Lewandowski, A.; Kopec, W.; Diakowski, W.; Chrzanowska, T. Oxidative DNA damage and total antioxidant status in serum of patients with esophageal squamous cell carcinoma. Hepatogastroenterology 2007, 54, 1701–1704. [Google Scholar] [PubMed]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer. 2008, 98, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. Ros-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E.; Ziegler, A.; Jonason, A.S.; Simon, J.A.; Kunala, S.; Leffell, D.J. Sunlight and sunburn in human skin cancer: P53, apoptosis, and tumor promotion. J. Investig. Dermatol. Symp. Proc. 1996, 1, 136–142. [Google Scholar] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006. [Google Scholar] [CrossRef]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Ponten, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C.; Hollstein, M. Clinical implications of the p53 tumor-suppressor gene. New Engl. J. Med. 1993, 329, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.M.; Loeb, L.A. Effect of DNA-repair enzymes on mutagenesis by oxygen free radicals. Mutat. Res. 1993, 289, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Levitt, N.C.; Hickson, I.D. Caretaker tumour suppressor genes that defend genome integrity. Trends Mol. Med. 2002, 8, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Olinski, R.; Jaruga, P.; Zastawny, T.H. Oxidative DNA base modifications as factors in carcinogenesis. Acta Biochim. Polonica 1998, 45, 561–572. [Google Scholar]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Hu, X.W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.H. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1alpha expression through activation of akt and p70s6k1 in human ovarian cancer cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, M.Z.; Azar, Z.M.; Srivastava, A.K. Role of receptor and nonreceptor protein tyrosine kinases in H2O2-induced PKB and ERK1/2 signaling. Cell Biochem. Biophys. 2007, 47, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, K. Carcinogen-mediated oxidant formation and oxidative DNA damage. Pharmacol. Ther. 1992, 53, 127–166. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Cantoni, O.; Tasinato, A.; Boscoboinik, D.; Azzi, A. Hydrogen peroxide-and fetal bovine serum-induced DNA synthesis in vascular smooth muscle cells: Positive and negative regulation by protein kinase C isoforms. Biochim. Biophys. Acta 1995, 1269, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the keap1-Nrf2-are pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Pantano, C.; Reynaert, N.L.; van der Vliet, A.; Janssen-Heininger, Y.M. Redox-sensitive kinases of the nuclear factor-kappab signaling pathway. Antioxid. Redox Signal. 2006, 8, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.D.; Eguchi, S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: Significance and involvement of egf receptor transactivation by angiotensin II. Antioxid. Redox Signal. 2003, 5, 771–780. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, F.R.; van Kranen, H.J.; Mullenders, L.H. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 19–27. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Trapp, V. Reactive oxygen species: An achilles’ heel of melanoma? Expert Rev. Anticancer Ther. 2008, 8, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.B.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Williams, S.M.G.; Reczek, E.E.; Johnson, B.W.; McGillicuddy, L.T.; Johannessen, C.M.; Hollstein, P.E.; MacCollin, M.; Cichowski, K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006, 10, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.G.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.H.; Basile, G.; Acosta, M.; Scott, C.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereirasmith, O.; et al. A biomarker that identifies senescent human-cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Yi, Q.F.; Travers, J.B.; Spandau, D.F. Uvb-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, K.; Deruy, E.; Martien, S.; Vercamer, C.; Bouali, F.; Dujardin, T.; Slomianny, C.; Houel-Renault, L.; Chelli, F.; de Launoit, Y.; et al. Senescent keratinocytes die by autophagic programmed cell death. Am. J. Pathol. 2009, 174, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Deruy, E.; Nassour, J.; Martin, N.; Vercamer, C.; Malaquin, N.; Bertout, J.; Chelli, F.; Pourtier, A.; Pluquet, O.; Abbadie, C. Level of macroautophagy drives senescent keratinocytes into cell death or neoplastic evasion. Cell Death Dis. 2014, 5, e1577. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres and telomerase: Their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (sips) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic RAS provokes premature cell senescence associated with accumulation of p53 and p16(Ink4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Jones, C.; Haughton, M.; DeMicco, C.; Kipling, D.; Wynford-Thomas, D. Direct evidence from siRNA-directed “knock down” that p16(Ink4a) is required for human fibroblast senescence and for limiting RAS-induced epithelial cell proliferation. Exp. Cell Res. 2004, 292, 151–156. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Starborg, M.; Brookes, S.; Peters, G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 1998, 8, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Maehara, K.; Yamakoshi, K.; Ohtani, N.; Kubo, Y.; Takahashi, A.; Arase, S.; Jones, N.; Hara, E. Reduction of total E2F/DP activity induces senescence-like cell cycle arrest in cancer cells lacking functional PRB and p53. J. Cell Biol. 2005, 168, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Udayakumar, T.; Shareef, M.M.; Diaz, D.A.; Ahmed, M.M.; Pollack, A. The E2F1/RB and p53/MDM2 pathways in DNA repair and apoptosis: Understanding the crosstalk to develop novel strategies for prostate cancer radiotherapy. Semin. Radiat. Oncol. 2010, 20, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16(Ink4a) tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Meloche, S.; Pouyssegur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO. J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.B.; Gish, K.; Murphy, M.; Yin, Y.X.; Notterman, D.; Hoffman, W.H.; Tom, E.; Mack, D.H.; Levine, A.J. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Gene Dev. 2000, 14, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Gene Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Y.; Porter, N.A. New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid. Redox Signal. 2005, 7, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.J.; Heck, D.E.; Mishin, V.; Black, A.T.; Shakarjian, M.P.; Kong, A.N.T.; Laskin, D.L.; Laskin, J.D. Modulation of keratinocyte expression of antioxidants by 4-hydroxynonenal, a lipid peroxidation end product. Toxicol. Appl. Pharmacol. 2014, 275, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.; Milkovic, L.; Zarkovic, N.; Waeg, G.; Rattan, S.I.S. Lipid peroxidation-derived 4-hydroxynonenal-modified proteins accumulate in human facial skin fibroblasts during ageing in vitro. Biogerontology 2014, 15, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Barland, O.C.; Ghadially, R.; Elias, P.M. Permeability and antioxidant barriers in aged epidermis. In Skin Aging; Springer: Berlin/Heidelberg, Germany, 2006; pp. 65–79. [Google Scholar]
- Shindo, Y.; Witt, E.; Packer, L. Antioxidant defense mechanisms in murine epidermis and dermis and their responses to ultraviolet light. J. Investig. Dermatol. 1993, 100, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Witt, E.; Han, D.; Tzeng, B.; Aziz, T.; Nguyen, L.; Packer, L. Recovery of antioxidants and reduction in lipid hydroperoxides in murine epidermis and dermis after acute ultraviolet radiation exposure. Photodermatol. Photoimmunol. Photomed. 1994, 10, 183–191. [Google Scholar] [PubMed]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545-589. https://doi.org/10.3390/biom5020545
Rinnerthaler M, Bischof J, Streubel MK, Trost A, Richter K. Oxidative Stress in Aging Human Skin. Biomolecules. 2015; 5(2):545-589. https://doi.org/10.3390/biom5020545
Chicago/Turabian StyleRinnerthaler, Mark, Johannes Bischof, Maria Karolin Streubel, Andrea Trost, and Klaus Richter. 2015. "Oxidative Stress in Aging Human Skin" Biomolecules 5, no. 2: 545-589. https://doi.org/10.3390/biom5020545
APA StyleRinnerthaler, M., Bischof, J., Streubel, M. K., Trost, A., & Richter, K. (2015). Oxidative Stress in Aging Human Skin. Biomolecules, 5(2), 545-589. https://doi.org/10.3390/biom5020545