1. Introduction
Infectious agents, such as foodborne pathogens, have caused numerous large-scale and costly outbreaks in the human history and will continue to be a major public health threat and financial burden for our society [
1,
2,
3,
4]. Early detection of pathogens, as the first step to prevent such outbreaks, has become increasingly more important today because the globalization of commerce and speedy travel have significantly increased the rate and breadth of the spread of infectious agents. Thus, the demand for faster, simpler, less expensive and more reliable pathogen testing methods has become ever greater.
Although the traditional culture method continues to be the ‘gold standard’ for bacterial detection, it is time-consuming and requires days or even weeks to complete (depending on the specific pathogen in question) [
5]. Modern methods take advantage of well-established biomolecular techniques, such as polymerase chain reaction (PCR) and immunoassay (where an antibody is used as molecular recognition element), to achieve faster and more sensitive pathogen detection [
5,
6,
7,
8,
9,
10,
11]. Despite the popularity of these techniques, they also come with certain drawbacks, such as the need for costly instrumentation and highly trained personnel to isolate or purify relevant targets (DNA for PCR and proteins for immunoassays). Thus, the entire test using such methods often still needs one or more days to complete. Detection sensitivity (for immunoassay) and tendency to generate false-positive results (for PCR) are also issues of concerns. For these considerations, we recently began to examine the utility of RNA-cleaving fluorogenic DNAzyme (RFD) probes for bacterial detection [
12,
13,
14]. RFDs can be isolated from random-sequence DNA pools to perform three linked functions: ligand binding, catalysis and fluorescence generation. Each RFD cleaves a synthetic nucleic acid substrate containing a single ribonucleotide as the cleavage site embedded in a DNA sequence, and the cleavage site is located between two nucleotides modified with a matching pair of fluorophore and quencher [
12,
13,
14,
15,
16,
17,
18,
19,
20,
21]. Because of these two features, these reporter molecules emit an increasing level of fluorescence when they carry out the catalytic cleavage of the RNA linkage. In other words, the cleavage event results in separation of the fluorophore from the quencher, accompanied by the increase of fluorescence intensity in real time.
More recently, we developed a method of isolating novel DNAzyme probes against the crude extracellular mixture (CEM) left behind by a specific type of bacteria in their environment or in the media they are cultured [
12]. The CEM is rich in diverse targets, including small molecules and proteins. Thus the use of the crude mixture as the complex target to conduct
in vitro selection [
22,
23,
24] experiment circumvents the tedious process of purifying and identifying a suitable target from the microbe of interest for biosensor development, and provides a subsequent assaying procedure that is simple because it does not require steps to purify a target of interest. Using this approach, we have isolated an RFD that cleaves its substrate only in the presence of the CEM produced by
E. coli (CEM-EC) [
12]. This
E. coli-sensing RFD, named RFD-EC1, was found to be highly selective to CEM-EC but nonresponsive to CEMs from many other Gram-negative and Gram-positive bacteria. We have also shown that the DNAzyme-based assay is capable of reporting the presence of a single
E. coli cell after 12 h of culturing. These experiments have illustrated the utility of RFDs as fluorogenic bacterial indicators. In this work we carried out a thorough investigation to characterize this bacterial detection system with a goal to further improve the detection sensitivity.
3. Experimental Section
3.1. Synthesis and Purification of Oligonucleotides
The standard DNA oligonucleotides (EC1, EC1T, EC1TM and EC1LT) were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA) and purified by 10% denaturing polyacrylamide gel electrophoresis (dPAGE). The modified oligonucleotide FS1 was acquired from W. M. Keck Oligonucleotide Synthesis Facilities (Yale University, New Haven, CT, USA), deprotected and purified by 10% dPAGE following a previously reported protocol [
15].
3.2. Enzymes and Chemical Reagents
T4 DNA ligase and T4 polynucleotide kinase (PNK) were purchased from MBI Fermentas (Burlington, ON, Canada). Tryptone and yeast extract was acquired from BD Biosciences (Mississauga, ON, Canada). All other chemical reagents were purchased from Sigma-Aldrich (Oakville, ON, Canada) and were used without further purification.
3.4. Preparation of Cis-Acting RFD-EC1
RFD-EC1 was generated by template-mediated ligation of FS1 to EC1. In brief, 200 pmol of FS1 were treated with 1× PNK buffer A (MBI Fermentas), 1 mM ATP and 20 U (units) of PNK for 30 min at 37 °C (reaction volume = 50 μL). The reaction was quenched by heating at 90 °C for 5 min. Equimolar EC1 and EC1LT (5’-CTAGG AAGAG TCGGA CGGAG CTG; the ligation template) were then added to this solution and was heated at 90 °C for 30 s and cooled to room temperature for 10 min. Afterwards, 10 μL of 10× T4 DNA ligase buffer (MBI Fermentas), 39 μL of deionized distilled water (ddH2O) and 1 μL of T4 DNA ligase (10 U/μL) were added. After incubation at room temperature (RT) for 2 h, the ligated EC1-FS1 was purified by 10% dPAGE.
3.5. Bacterial Cells
Gram-negative bacteria Pseudomonas peli, Yersinia rukeri, Hafnea alvei, and Achromobacter xylosoxidans were donated by Dr. Gerard Wright (Micheal G. DeGroote Institute for Infectious Disease Research, McMaster University). Gram-positive bacteria Leuconostoc mesenteroides, Lactobacillus planturum and Pediococcus acidilactici (PA) were gifts from Dr. Brian Coombes and Dr. Russel Bishop (Department of Biochemistry and Biomedical Sciences, McMaster University). E. coli K12 (MG1655) and Bacillus subtilis 168 are regularly maintained in our laboratory.
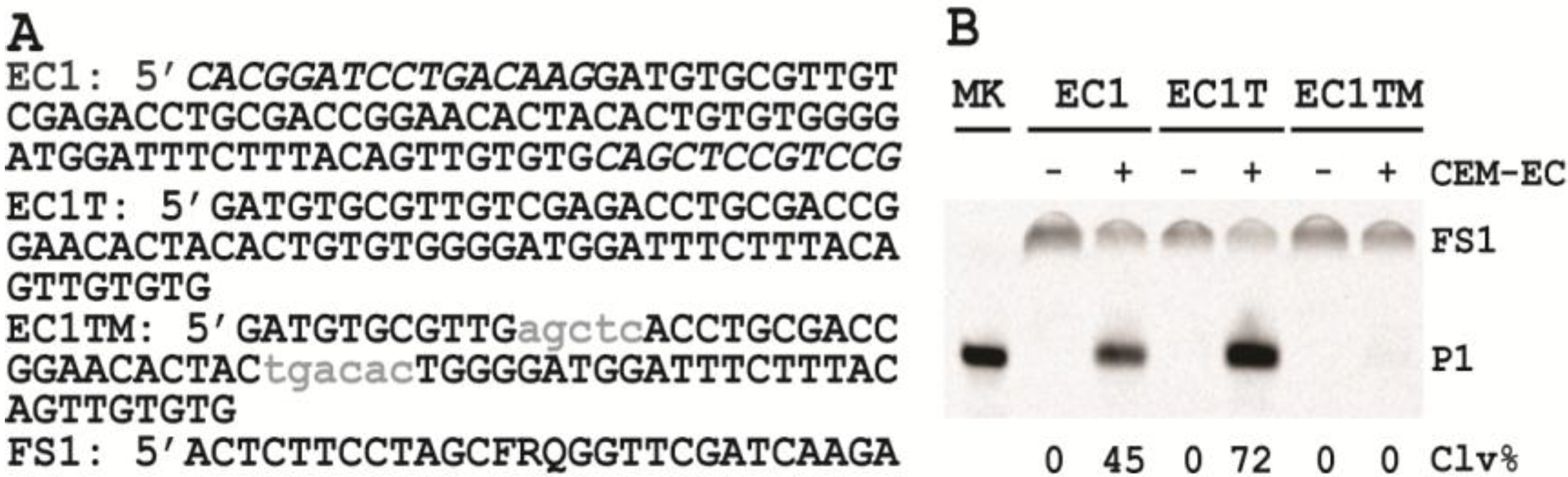
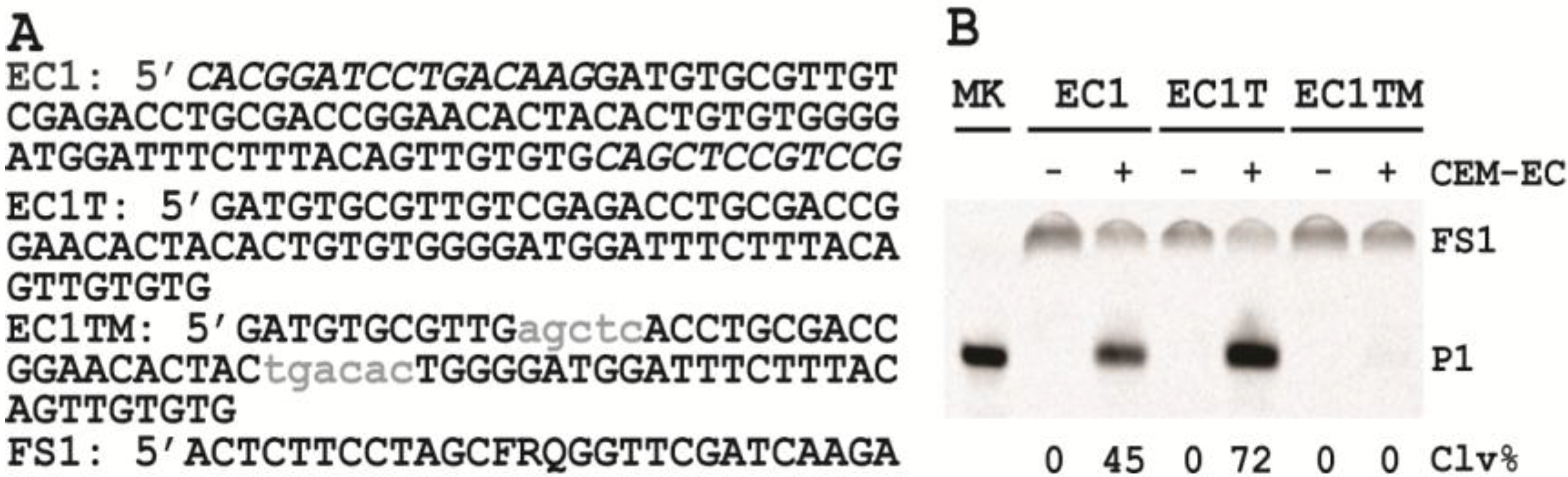
3.6. Comparison of the Cleavage Activity of EC1, EC1T and EC1TM in the Presence of CEM-EC
E. coli was plated onto a TSB agar (1.5%) plate and grown for 14 h at 37 °C. A single colony was taken and inoculated into 2 mL of TSB and grown for 14 h at 37 °C with shaking at 250 rpm. A 1% fresh culture was made by re-inoculating 20 µL of the above culture into 2 mL of TSB. The re-inoculation was allowed to grow at 37 °C with shaking at 250 rpm until the culture reached an OD600 of ~1. 1 mL of this culture was centrifuged at 11,000 g for 5 min at room temperature; the supernatant was taken as the crude extracellular mixture (CEM-EC) and stored at −20 °C.
For each candidate DNAzyme construct, two reactions were set up, a control and a test. For the test, 25 μL of 2× reaction buffer (2× RB; 100 mM HEPES, 300 mM NaCl, 30 mM MgCl2, pH 7.5) was mixed with 23 μL of the CEM-EC prepared above, 1 μL of 2.5 μM FS1 and 1 μL of 2.5 μM EC1, EC1T or EC1TM. For the control, TSB was used to substitute the CEM-EC. Each reaction mixture was incubated at RT for 60 min, followed by quenching with 5 μL of 3 M NaOAc (pH 5.5) and 135 μL of cold ethanol. DNA was recovered by centrifugation and analyzed by 10% dPAGE. DNA bands in the gel were visualized by Typhoon 9200 (GE Healthcare) and quantified by ImageQuant software (Molecular Dynamics).
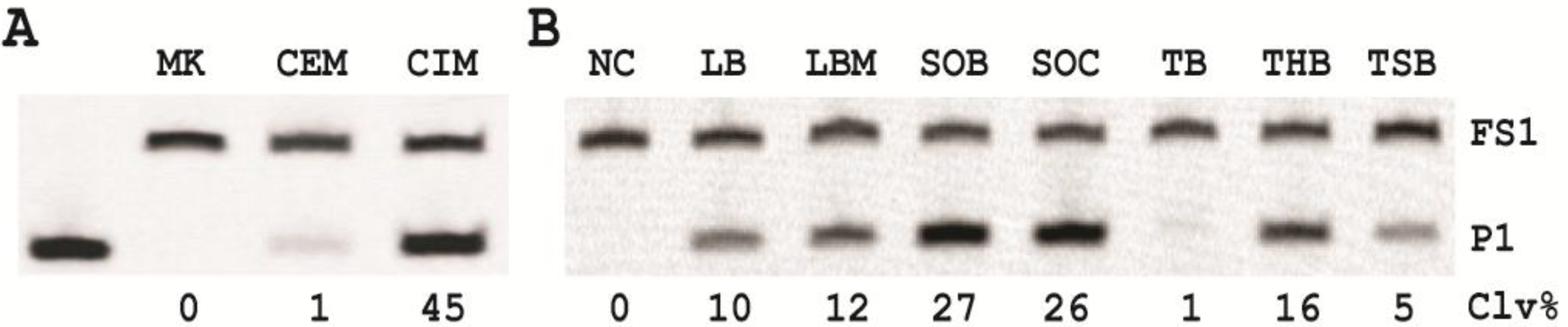
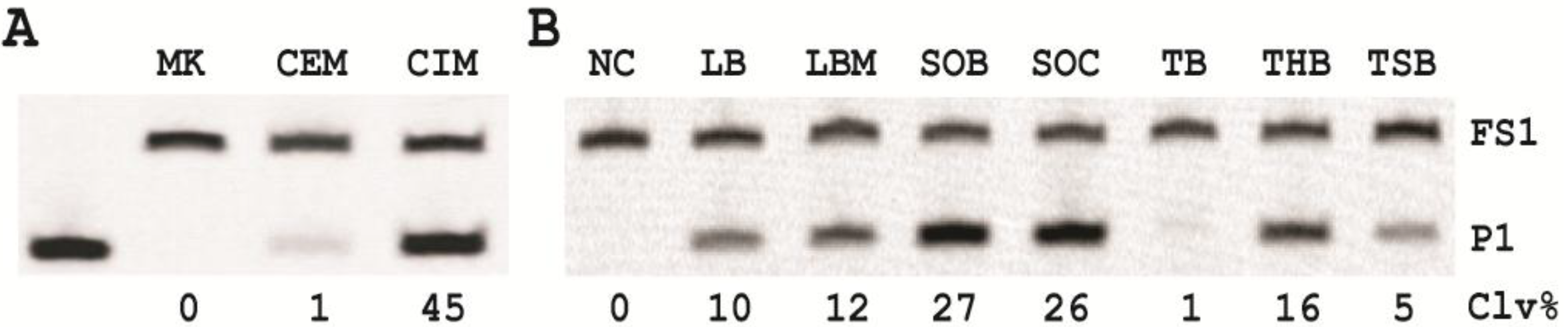
3.7. Comparison of the Cleavage Activity of EC1T in the Presence of CEM-EC and CIM-EC
100 μL of 50,000 CFU/mL glycerol stock of E. coli was inoculated into 2 mL of TSB and grown at 37 °C for 7 h with shaking at 250 rpm. 1 mL of this culture was centrifuged at 11,000 g for 5 min at room temperature; the supernatant was taken as the CEM-EC for this experiment. The cell pellet was suspended in 200 µL of 1× RB and heated at 50 °C for 15 min. The heat-treated cell suspension was then centrifuged at 11,000 g for 5 min at RT. The clear supernatant was taken as the CIM-EC for the experiment.
The cleavage reaction with the CEM-EC was carried out by mixing 25 μL of 2× RB, 23 μL of the CEM-EC prepared above, 1 μL of 2.5 μM FS1 and 1 μL of 2.5 μM EC1T. The reaction concerning the CIM-EC was conducted by mixing 41 μL of 1× RB, 5 μL of CIM-EC, 1 μL of 2.5 μM FS1, 1 μL of 2.5 μM EC1T and 2 μL of 2× RB (note that the CIM-EC was made by suspending the cell pellet from originally 1 mL of E. coli culture in 200 μL of 1× RB, which translates into a concentrating factor of 5). A control experiment without the CEM-EC and CIM-EC was also conducted. Each reaction mixture was incubated at RT for 60 min, followed by 10% dPAGE analysis as described above.
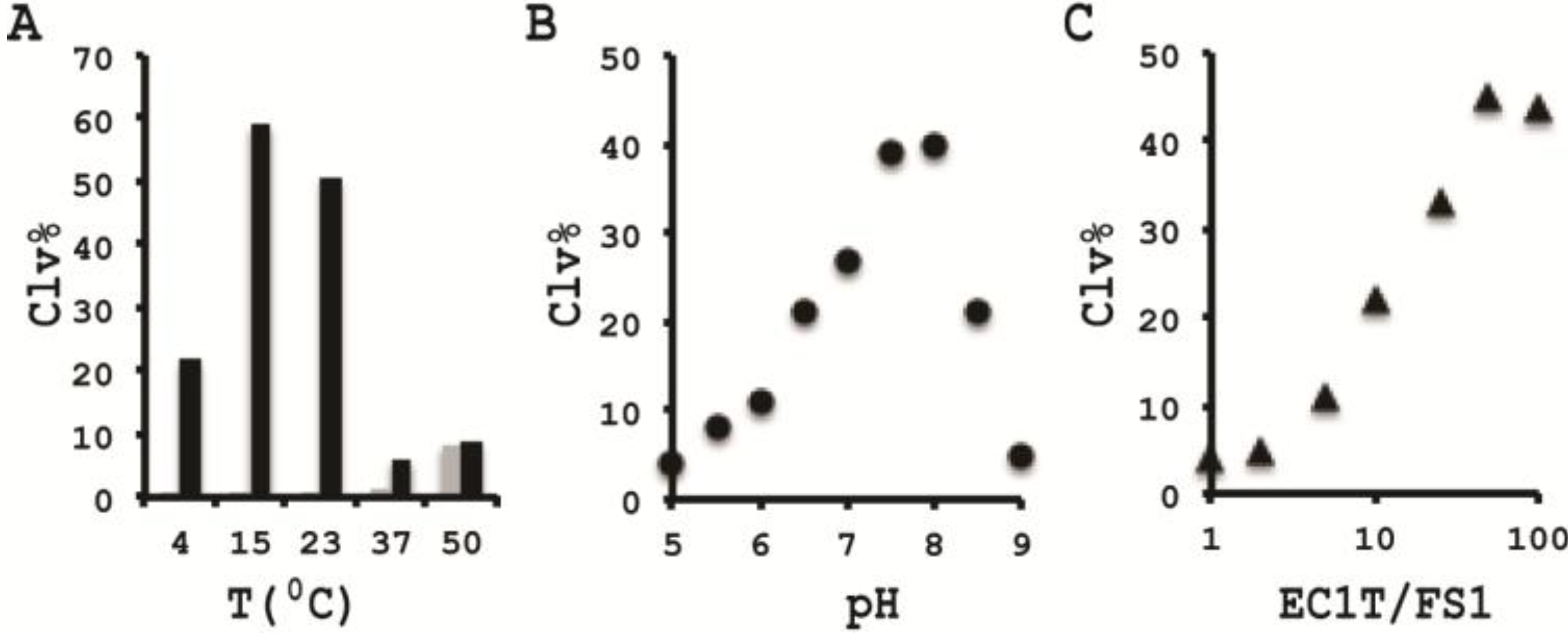
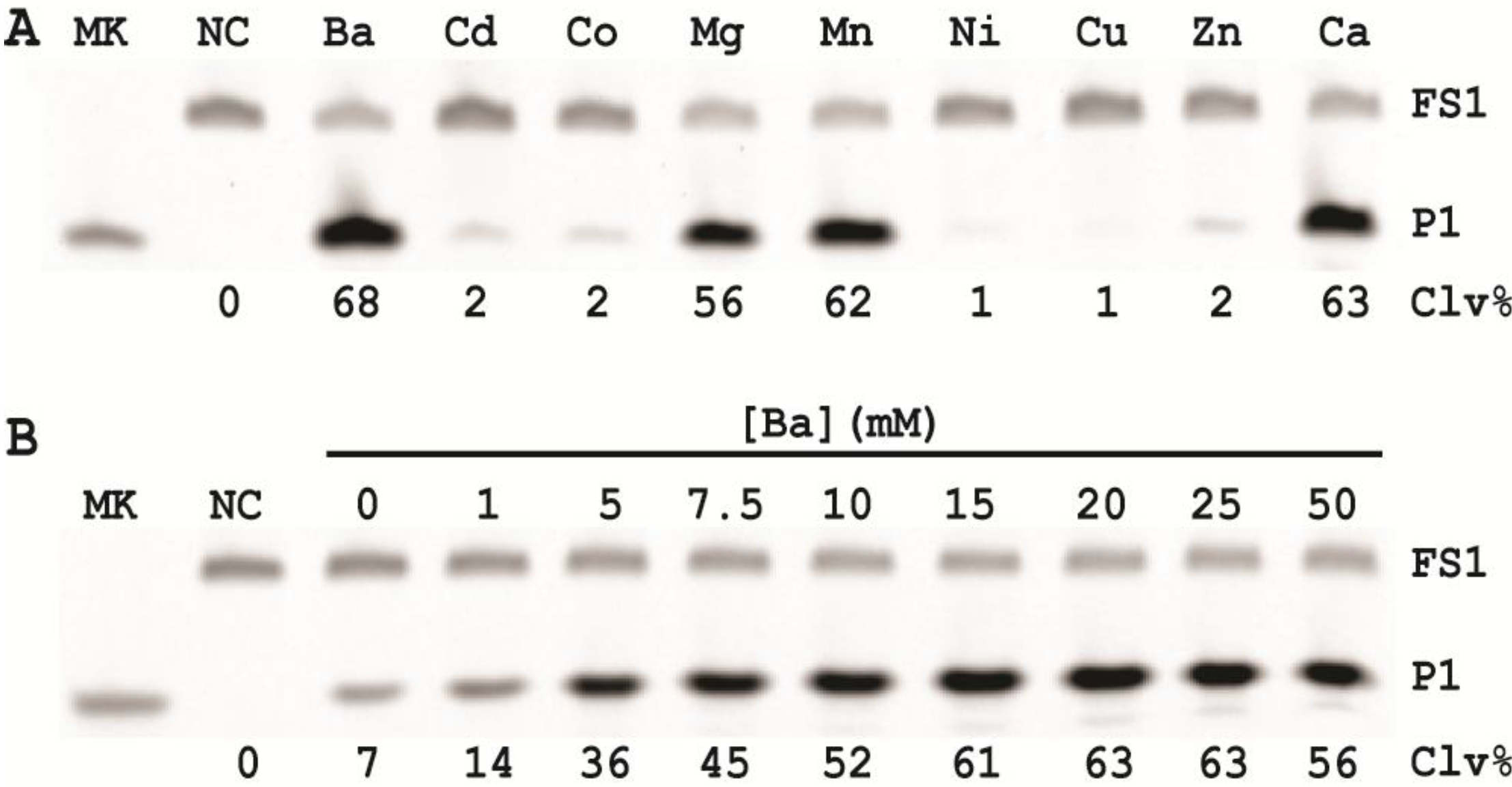
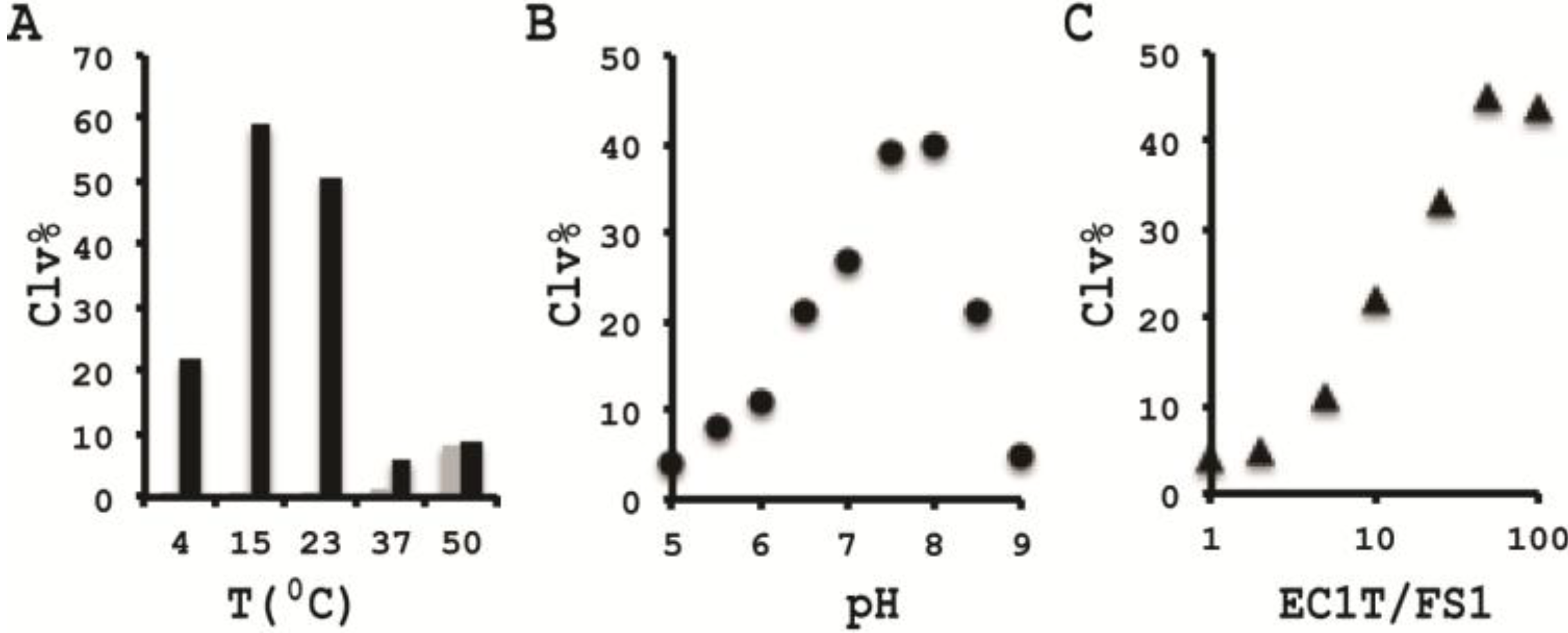
3.10. Comparison of the Cleavage Activity of EC1T at Different Reaction Temperature
A 2× RBBa stock (100 mM HEPES, 300 mM NaCl, 30 mM BaCl2, pH 7.5) was first prepared. Five cleavage reaction mixtures were then set up by mixing 19.5 μL of water, 22.5 μL of 2× RBBa, 1 μL of 2.5 μM FS1, 1 μL of 2.5 μM EC1T, and 5 μL of the CIM-EC prepared with 1× RBBa. These mixtures were incubated, respectively, at 4, 15, 23, 37 and 50 °C for 60 min, followed by 10% dPAGE analysis as described above.
3.11. Comparison of the Cleavage Activity of EC1T at Different pH
A series of 2× RBBaʹ stock (300 mM NaCl, 30 mM BaCl2, along with a chosen buffering agent at 100 mM) were first prepared with pH being varied from 5.0 to 9.0 at an increasing interval of 0.5 units. MES was used for pH 5.0, 5.5 and 6.0; HEPES was used for pH 6.5, 7.0, 7.5 and 8.0; Tris was used for pH 8.5 and 9.0. The cleavage reactions were then conducted in a similar fashion as described in the section immediately above. Note that the CIM-EC for a given pH was prepared with a relevant 1× RBBaʹ.
3.12. Comparison of the Cleavage Activity of EC1T at Varying FS1/EC1T Ratios
Stocks of EC1T at 2.5, 5, 12.5, 25, 62.5, 125, and 250 μM were first prepared. Cleavage reactions were then conducted by mixing 19.5 μL of water, 22.5 μL of 2× RBBa, 1 μL of 2.5 μM FS1, 1 μL of a given EC1T stock, and 5 μL of the CIM-EC prepared with 1× RBBa. Each reaction mixture was incubated at RT for 60 min, followed by 10% dPAGE analysis as described above.
3.13. Specificity Test
Five Gram-negative bacteria (P. peli, Y. rukeri, H. alvei, A. xylosoxidans and E. coli) and four Gram-positive bacteria (L. mesenteroides, L. planturum, P. acidilactici and B. subtilis) were tested in this experiment. Each bacterium was cultured in SOB for a different period of time until the OD600 reached ~1. The CIM was then prepared with 1× RBBa and tested with EC1T/FS1 under the optimal reaction condition (50 mM HEPES, pH 7.5, 150 mM NaCl and 15 mM BaCl2, room temperature, EC1T/FS1 = 50/1). Each reaction mixture was incubated at RT for 60 min, followed by 10% dPAGE analysis as described above.
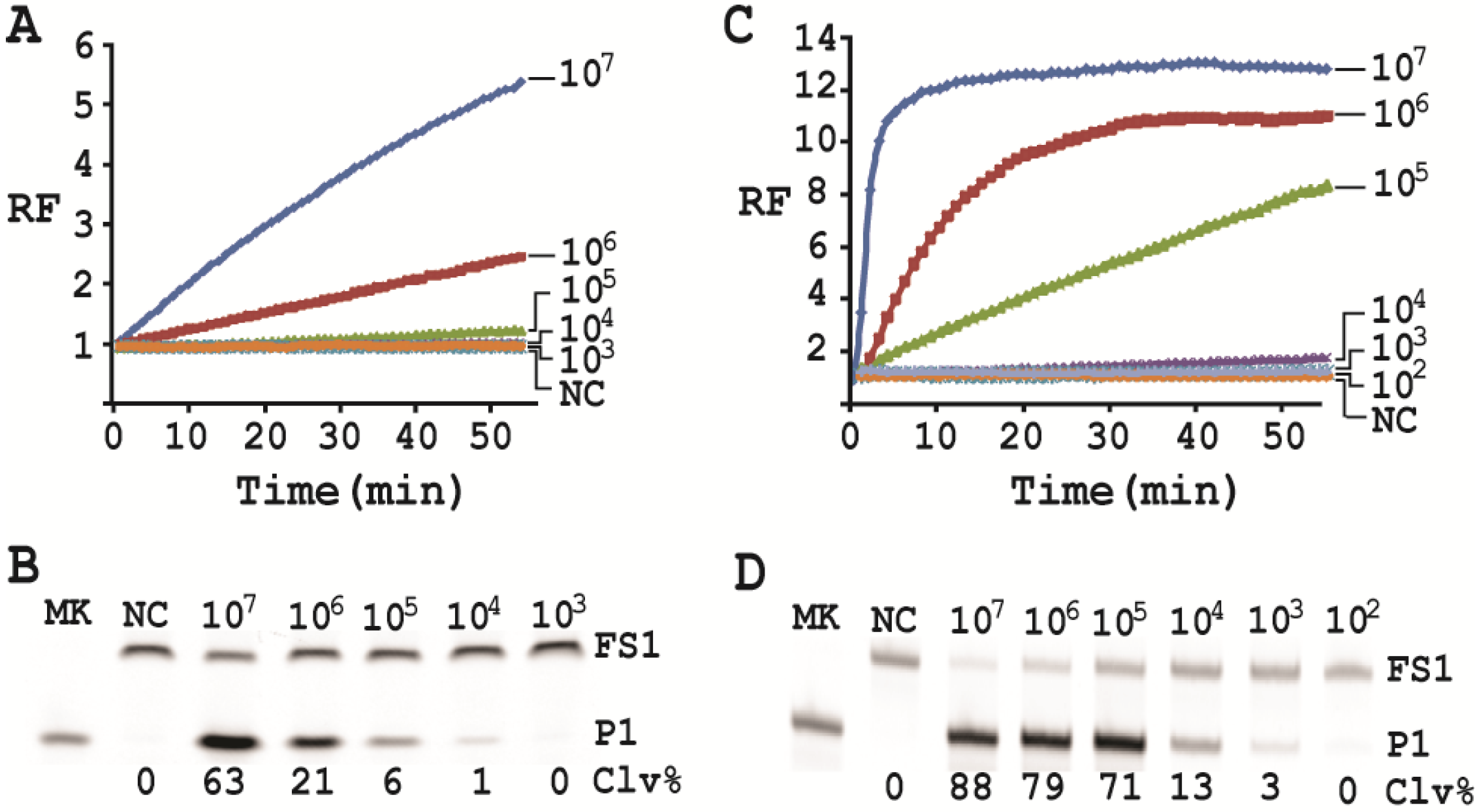
3.14. Detection Sensitivity
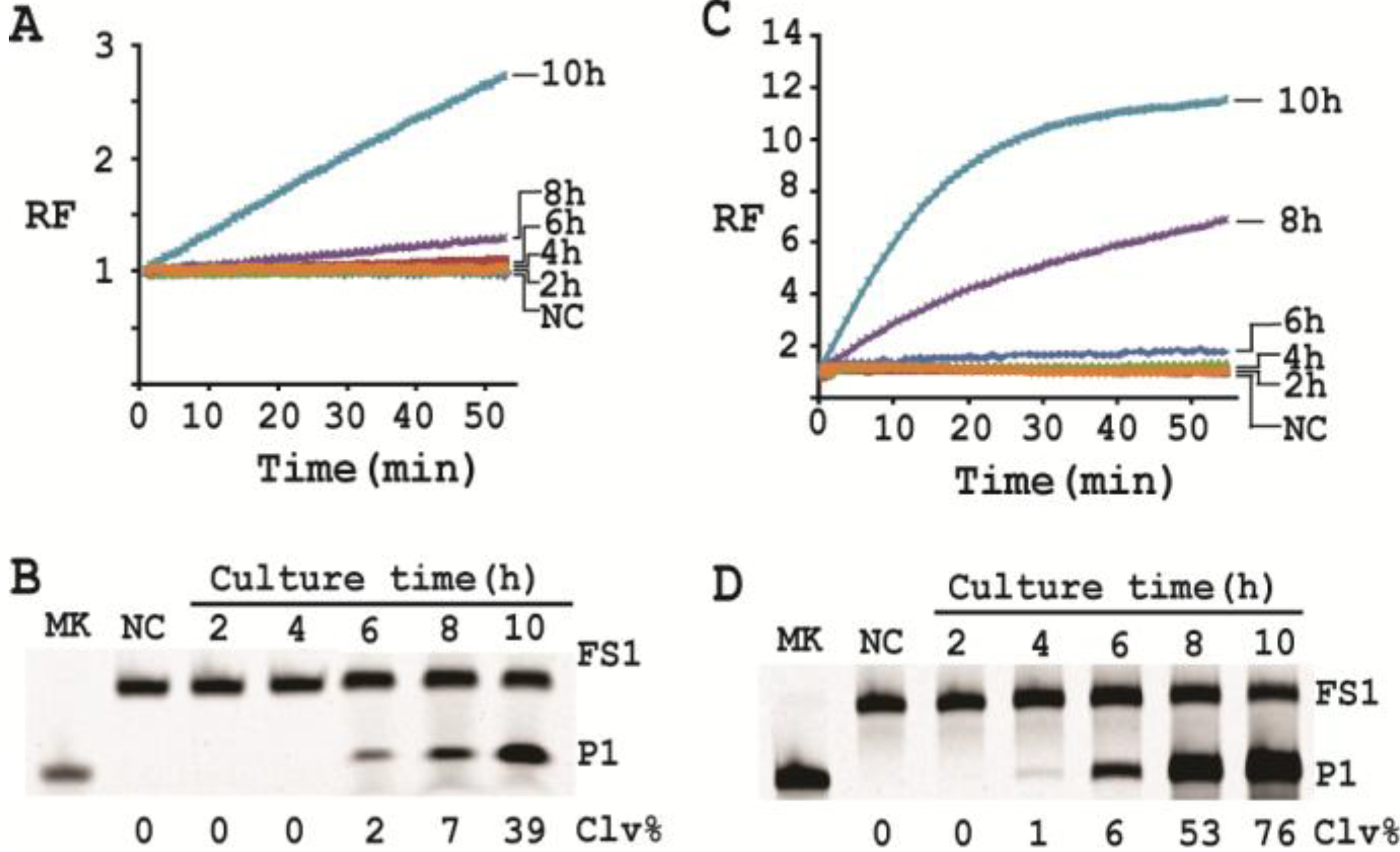
First, a single colony of E. coli from an agar plate was taken, inoculated into 2 mL of SOB and grown for 14 h at 37 °C with shaking at 250 rpm. 10-fold serial dilution was then carried out as follows: 100 μL of the 14-h culture was mixed with 900 μL of fresh SOB. 100 μL of the diluted culture was again taken and mixed with 900 μL of fresh SOB. This process was repeated 7 times. 100 µL of the final dilution were plated onto a TSB agar plate (done in triplicate), which was incubated at 37°C for 15 h. Colonies in each plate were counted; the average number of colonies from the three plates was taken as the number of cells for this final dilution. This number was then used to calculate the number of cells for the other dilutions. 500 μL of each dilution was taken and centrifuged at 11,000 g for 5 min at RT. The cell pellet was re-suspended in 100 μL of 1× RBBa and used as the CEM-EC for this experiment (done in triplicate).
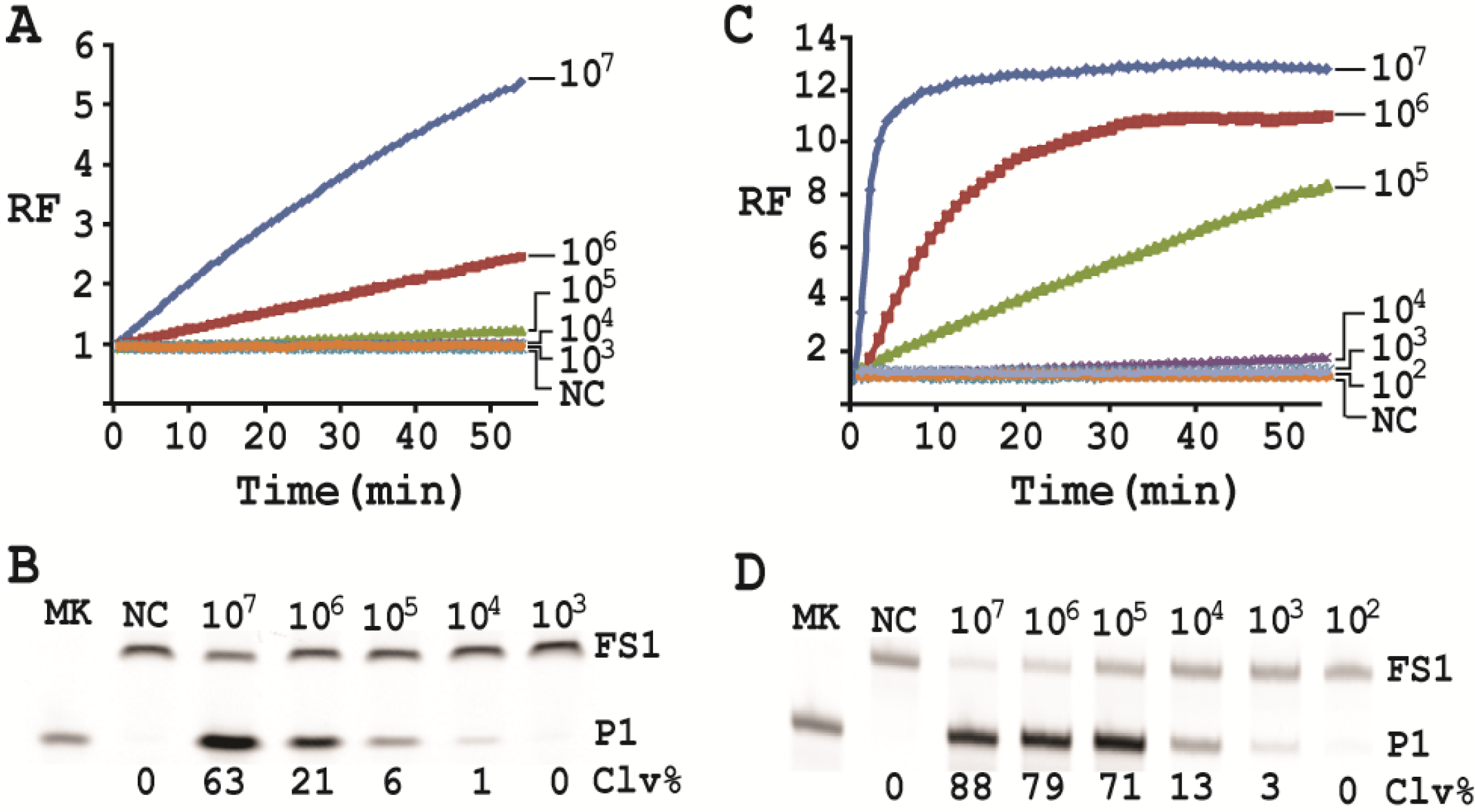
Cleavage reactions concerning EC1T/FS1 were set up and monitored as follows: 19.5 μL of water, 22.5 μL of 2× RB
Ba, 1 μL of 2.5 μM FS1, 1 μL of 125 μM EC1T were mixed in a quartz crystal cuvette, which was placed in a fluorimeter (Cary Eclipse Fluorescence Spectrophotometer; excitation wavelength = 488 nm and emission wavelength = 520 nm) set at RT. Fluorescence intensity was recorded every minute for 5 min; 5 μL of a relevant CIM-EC was then added into the cuvette and the solution was quickly mixed by pipetting the mixture up and down a few times. Following this step, the fluorescence intensity of the solution was recorded for 55 more minutes. All the reactions were conducted in 3 replicates and the average data are shown in
Figure 6A. The final reaction mixture was also taken and analyzed by 10% dPAGE and data are shown in
Figure 6B.
Cleavage reactions concerning RFD-EC1 were set up and monitored similarly: 20.5 μL of water, 22.5 μL of 2× RB
Ba, 1 μL of 2.5 μM RFD-EC1 was mixed in a cuvette. After reading fluorescence intensity for 5 min, 5 μL of a relevant CIM-EC was then added, followed by fluorescence intensity reading for 55 more minutes (
Figure 6C). The final reaction mixture was also analyzed by 10% dPAGE (
Figure 6D).
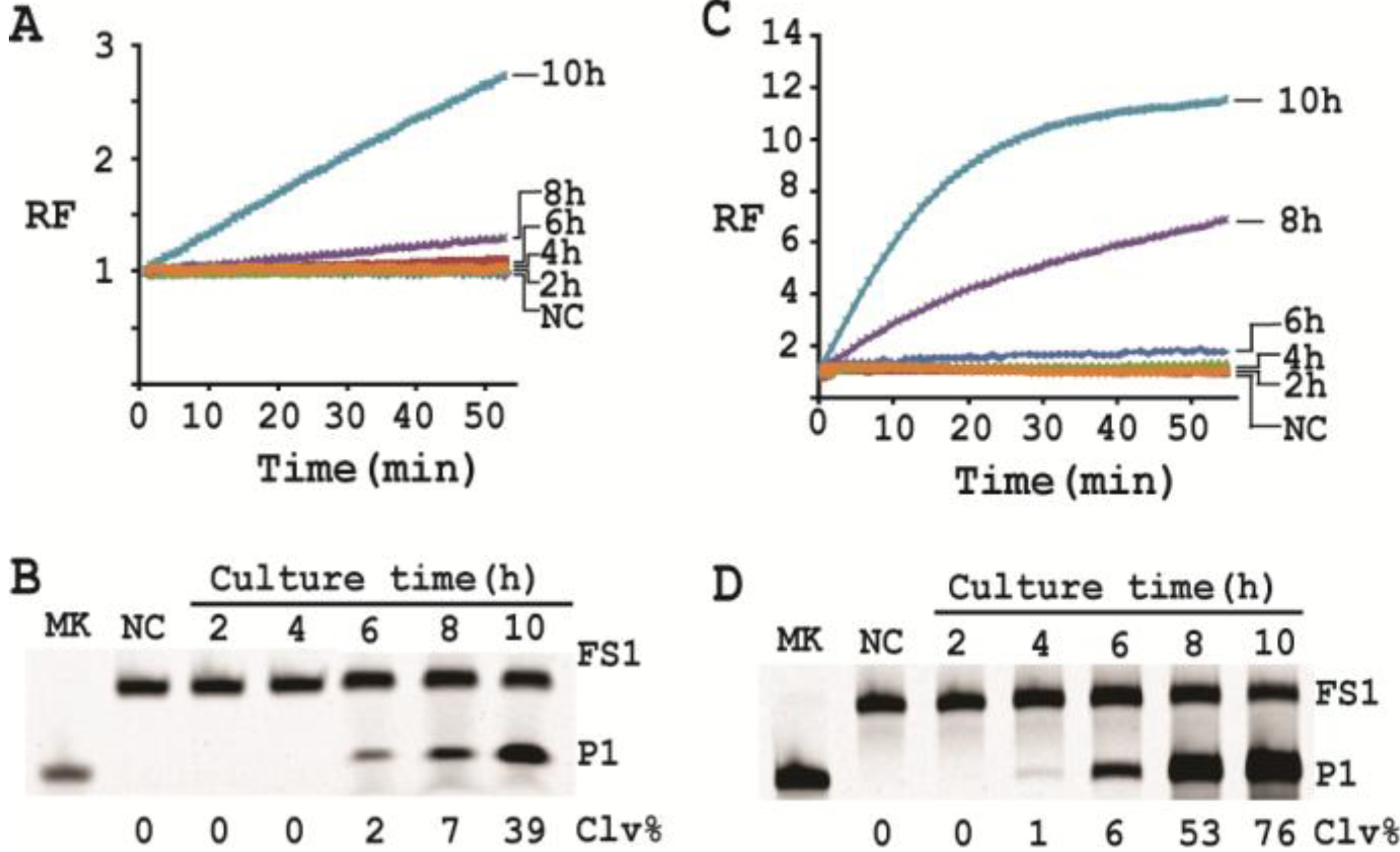
3.15. Single Cell Detection via Culturing
For isolating a single cell we followed our previously reported protocol [
12]. Briefly, a glycerol stock containing 2 CFU/mL of
E. coli was prepared. 100 μL of this stock was distributed to 10 culture tubes each with 2 mL of SOB. Since the concentration of the stock was 2 CFU/mL, only 2 out of the 10 tubes contained a single seeding cell (2 CFU/mL × 0.1 mL = 2). All the tubes were incubated at 37 °C with shaking at 250 rpm. At 2, 4, 6, 8 and 10 h, 200 μL of culture was harvested from each culture tube and CIMs were prepared (40 μL of 1× RB
Ba was used to dissolve the cell pellet). All the tubes were further incubated for 20 h to identify the two tubes containing
E. coli cell (the culture in these tubes turned turbid while that in other 8 tubes stayed clear). Each CIM from
E. coli-containing tubes was used to initiate the cleavage reaction by mixing 19.5 μL of water, 22.5 μL of 2× RB
Ba, 1 μL of 2.5 μM FS1, 1 μL of 125 μM EC1T, and 5 μL of a relevant CIM. The reaction and dPAGE analysis procedures were same as described above.
4. Conclusions
We recently described an RNA-cleaving fluorogenic DNAzyme, named RFD-EC1, which is active in the presence of the crude extracellular mixture (CEM) of the model Gram-negative bacterium
E. coli [
12,
13,
14]. RFD-EC1 was found to be highly active with CEM of
E. coli but inactive with CEMs from a host of other Gram-negative and Gram-positive bacteria, and thus, RFD-EC1 can be used to develop a simple, “mix-and-read” fluorescence assay to achieve selective detection of
E. coli. However, several parameters that are particularly relevant to the performance of this assay remained to be investigated. In this study we sought to establish a
trans-acting DNA catalyst that cleaves an external substrate, optimize the reaction conditions that best support the catalytic activity of the DNAzyme, and determine the culturing conditions that enable the quickest detection of a single live bacterial cell.
The trans-acting DNAzyme was successfully established by segregating the substrate sequence domain from the sequence of the original DNA library. Also the two fixed sequence domains flanking the random-sequence domain could be removed without affecting the catalytic performance of the DNAzyme. The shortened, trans-acting DNAzyme, named EC1T, now contains 70 nucleotides.
Originally, the DNAzyme was isolated to cleave in the presence of the crude extracellular mixture (CEM) of
E. coli and it has been determined that the target that activates the DNAzyme is a protein molecule based on the observation that the treatment of the CEM with proteases abolishes the DNAzyme activity [
12]. Although the identity of this target is yet to be determined, we found that the target protein is much more abundant intracellularly and could be retrieved with a simple heating step (50 °C; 15 minutes). This led us to the use of the crude intracellular mixture (CIM) as the target of detection, translating into a better assay sensitivity.
Our results revealed that the nutritional factors in culture media played a vital role in growing the cells in faster rate (varying by as much as ~25-fold) with super Optimal Broth (SOB) which can substantially reduce the time required for single cell detection.
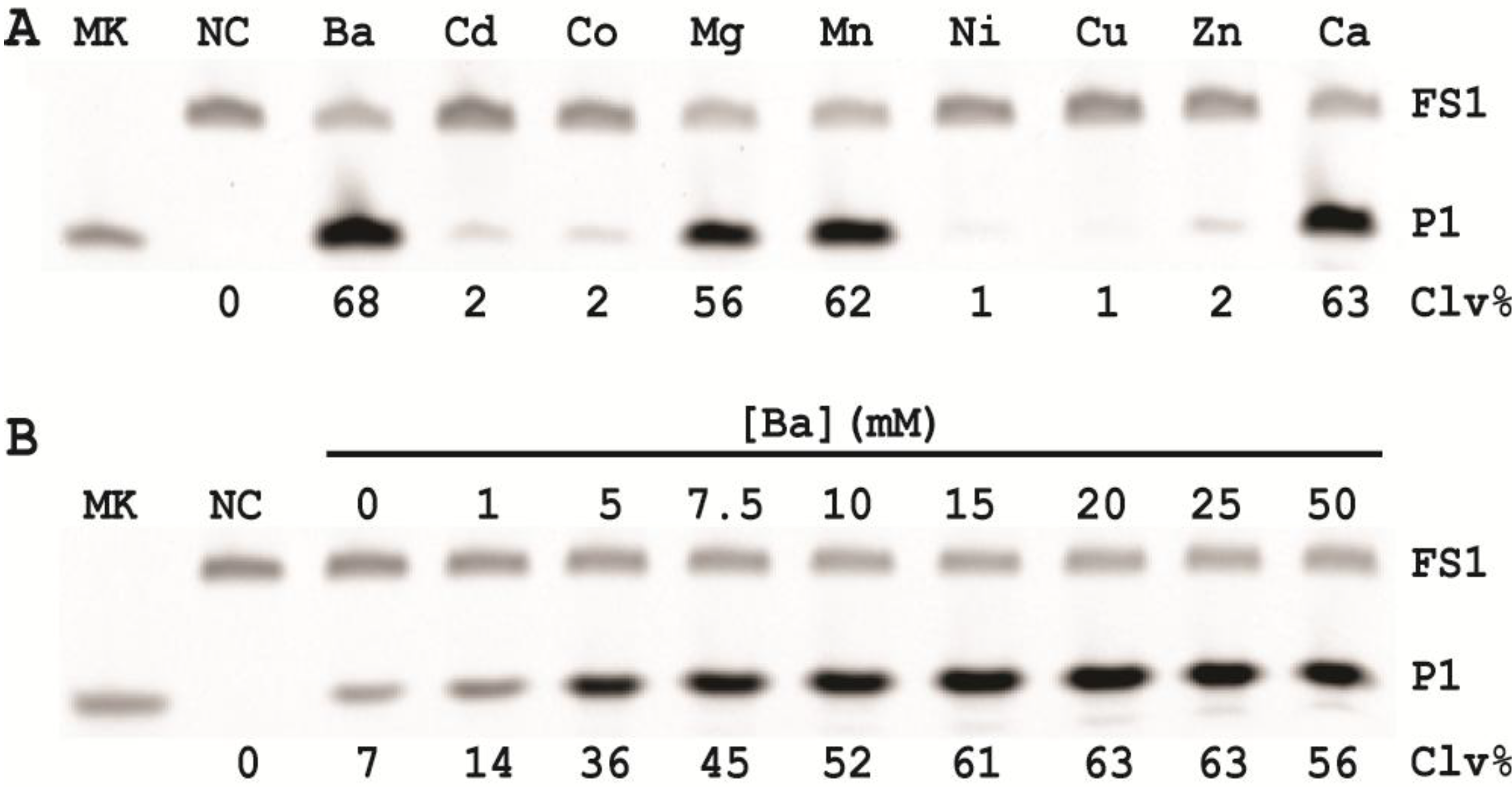
In order to establish an optimal reaction condition for EC1T, we examined the following reaction parameters: choice of divalent metal ions, reaction temperature and pH as well as the ratio between the substrate and the DNAzyme. Although EC1T was found to be active in the absence of any divalent ion, it exhibited much stronger activity in the presence of Ba2+, Ca2+, Mn2+ or Mg2+. We chose Ba2+ as the divalent metal ion cofactor because this metal ion does not impose any fluorescence quenching effect. The DNAzyme was originally derived at room temperature (~23 °C) and a solution pH of 7.5 and therefore it was not surprising that EC1T exhibited the strongest activity at 23 °C and pH 7.5. We further found that when the concentration of FS1 was kept at 50 nM, 2.5 μM EC1T was required to reach the optimal cleavage activity. All the above optimization experiments led to the establishment of the optimal reaction condition for EC1T: 50 mM HEPES, 150 mM NaCl, 15 mM BaCl2, pH 7.5, DNAzyme: substrate ratio = 50:1.
Under the above optimal reaction condition, the trans-acting system was able to detect 105 cells when the reaction was monitored in a fluorimeter. If the reaction mixture was analyzed by dPAGE (which separates the reaction product from the substrate), the system can detect 104 cells. When the original RFD-EC1 was used for the assay, the detection sensitivity was further improved: the fluorimeter method was able to detect 104 cells while the dPAGE method was able to detect as low as 103 cells. Importantly, the optimized assay did not compromise the specificity.
With a culturing step, the optimized assay is able to achieve the detection of E. coli from a single colony forming unit in 4–6 hours (dependent on the method of choice), which represents a significant deduction in time (12 h) required by the same probe under unoptimized conditions. Overall, we have significantly improved the performance of our DNAzyme probe and demonstrate the utility of such probes as simple biosensors to achieve sensitive and speedy detection of bacterial pathogens.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}