

Active Colitis-Induced Atrial Electrophysiological Remodeling
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Electrocardiogram Recordings
2.3. Isolated Langendorff Perfused Hearts
2.4. Current and Voltage Clamp Recordings
2.5. Isolation of Mouse Atrial Myocytes
2.6. Protein Quantification by Western Blot
2.7. Chemicals
2.8. Statistic
3. Results
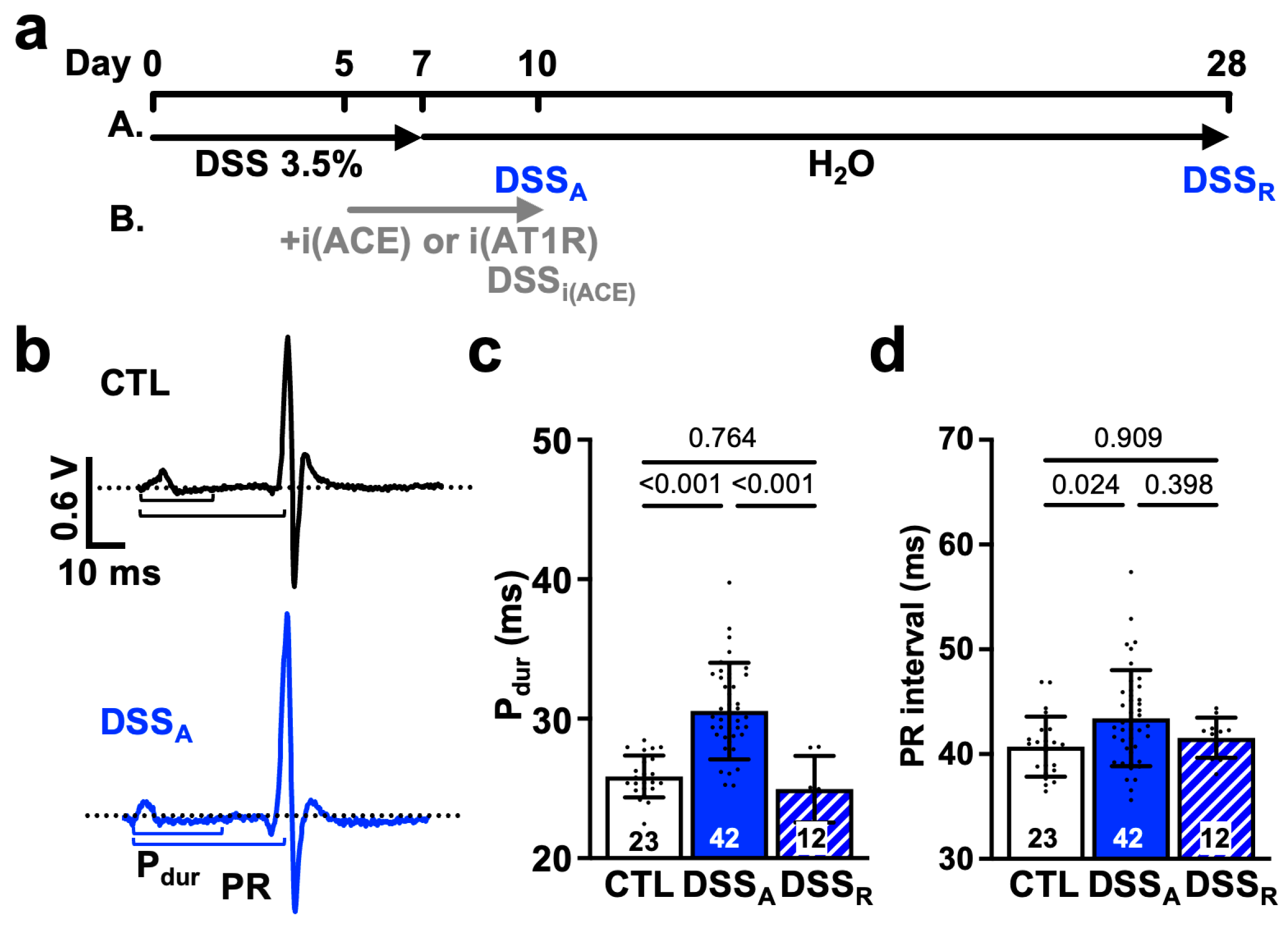
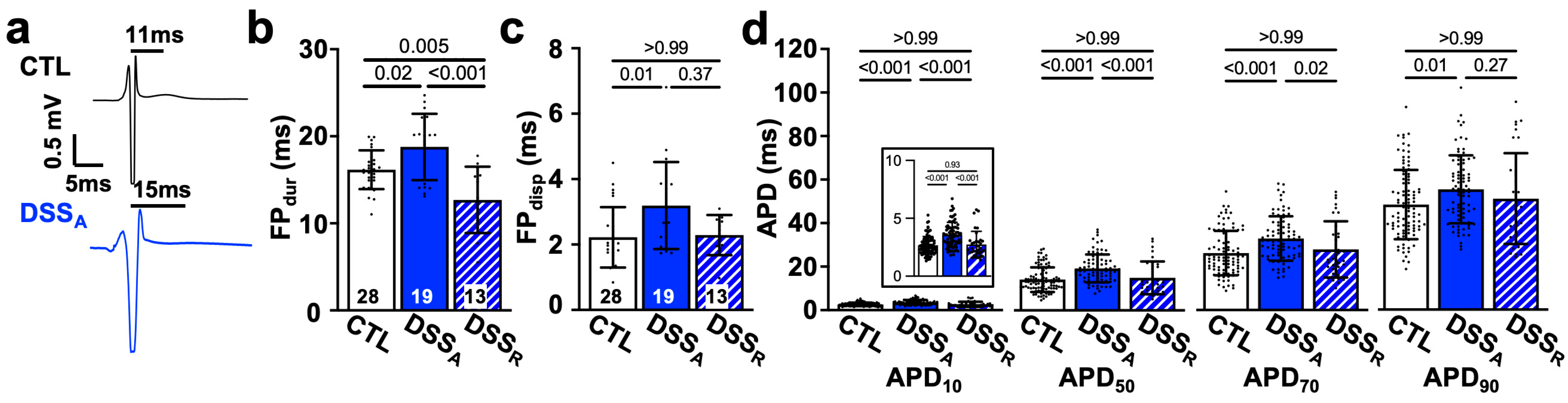
3.1. Change in Atrial Electrophysiological Properties During Active Colitis
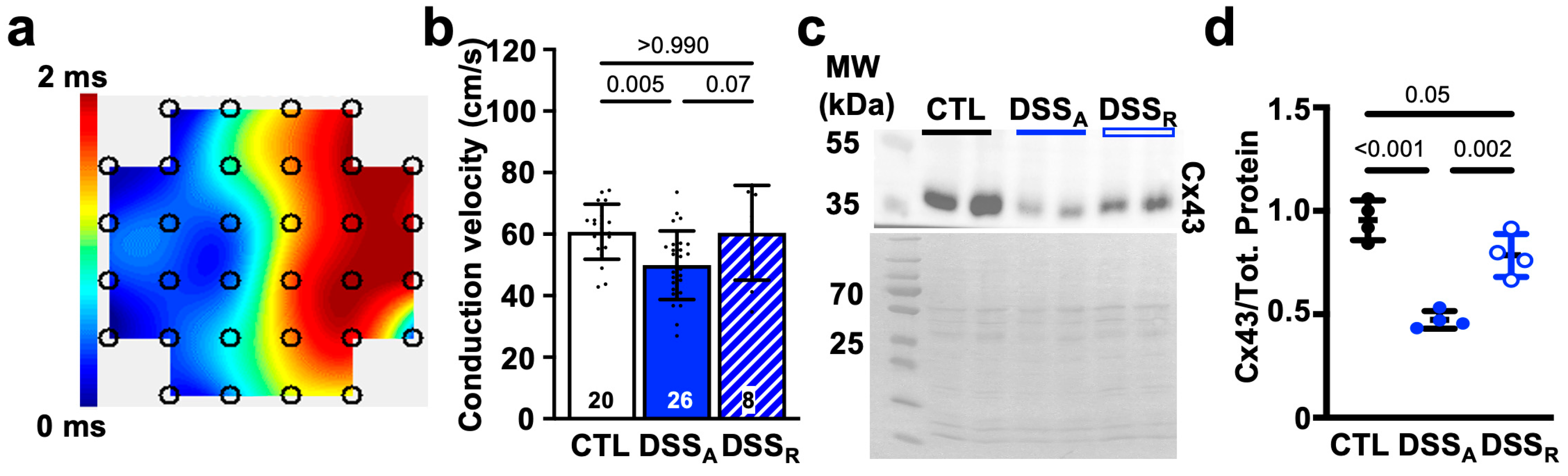
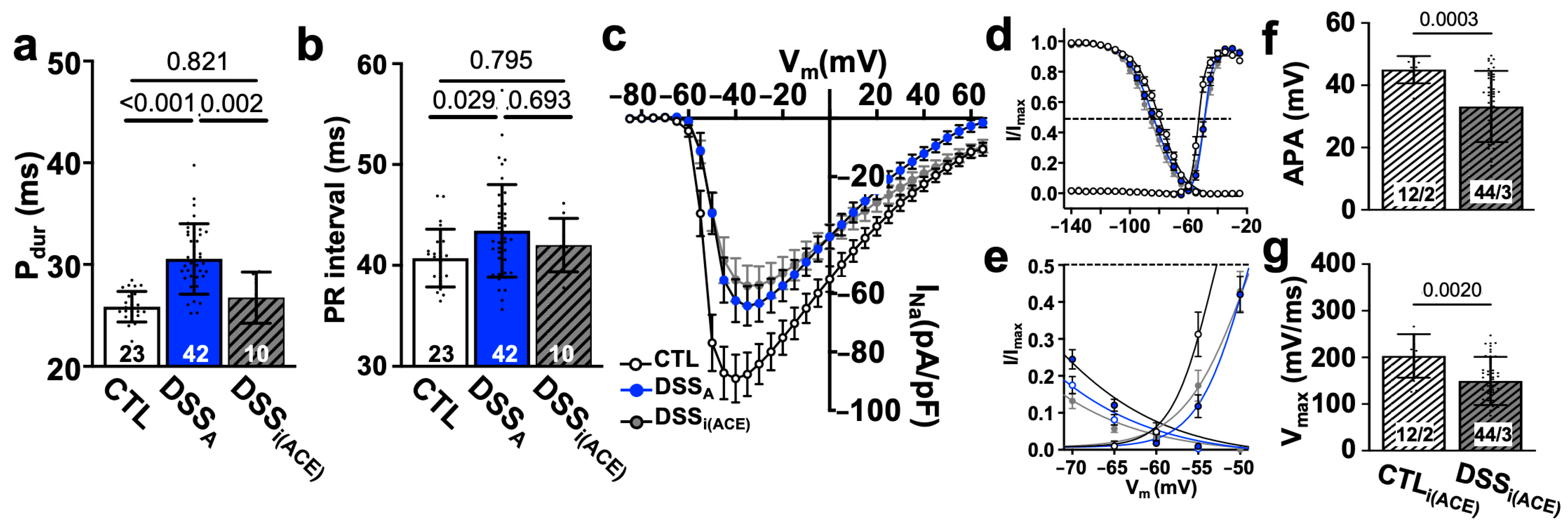
3.2. Colitis-Induced Changes of the Atrial Conduction Velocity
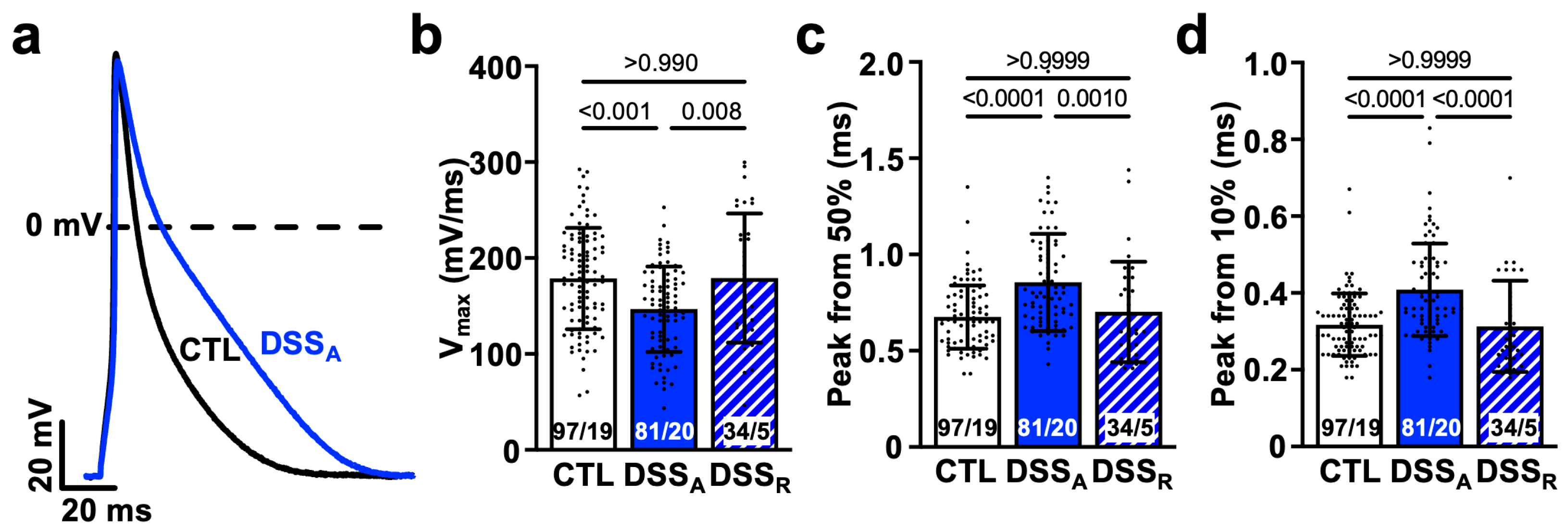
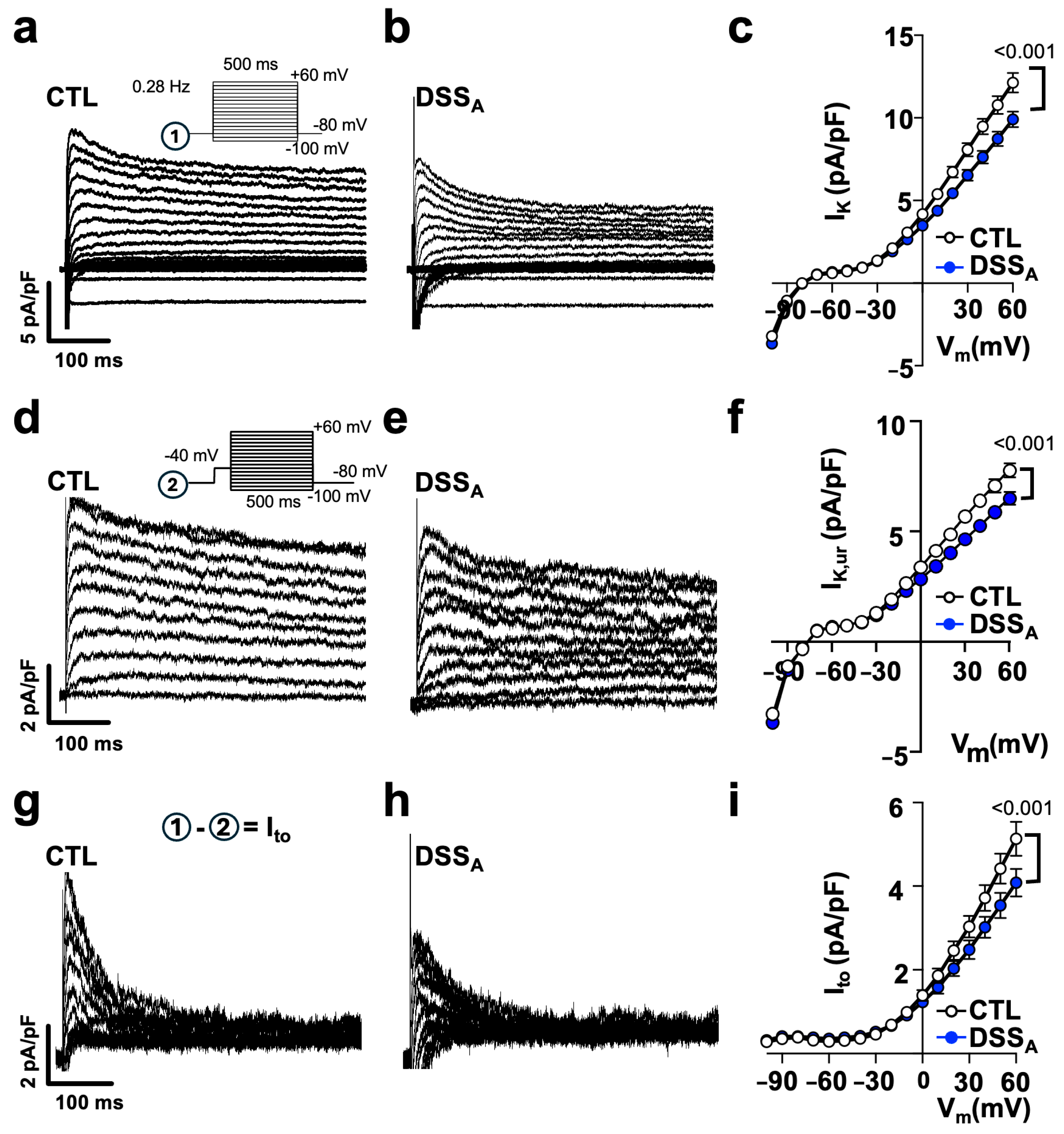
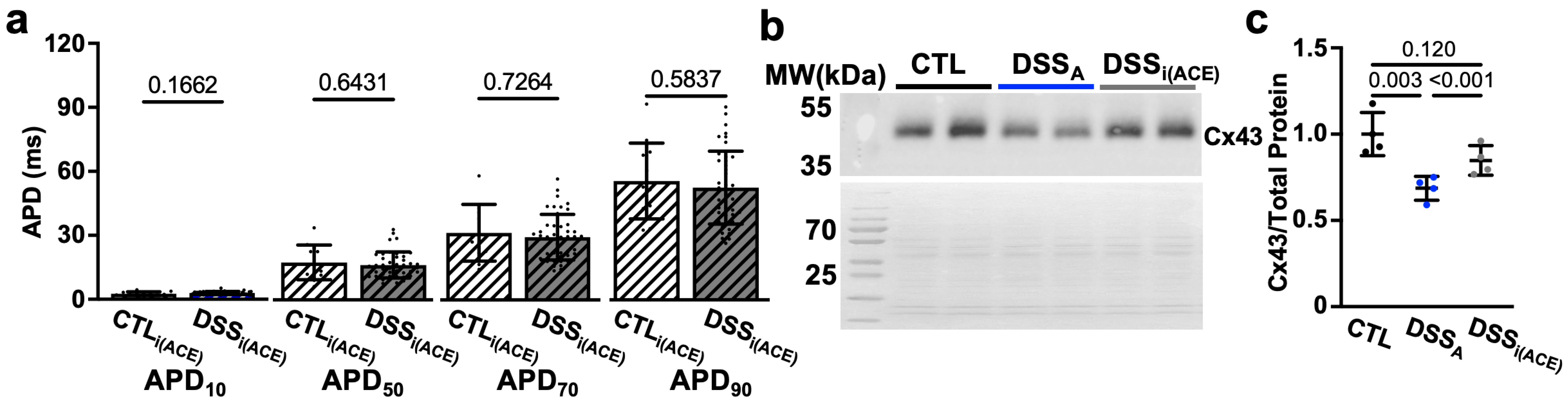
3.3. Colitis-Induced Changes of the Atrial APD
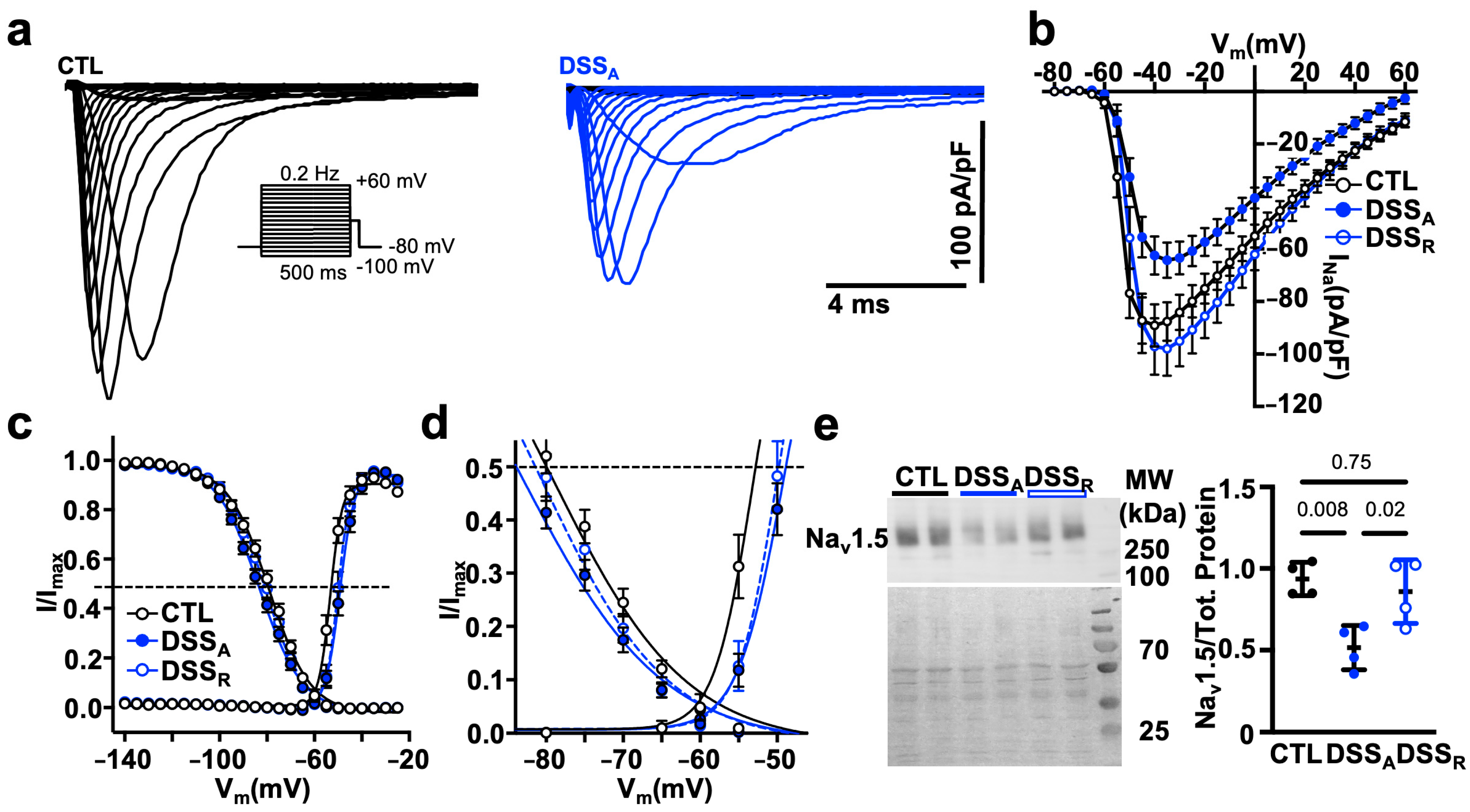
3.4. Increased RAS Signaling Links Colitis to Atrial Remodeling
4. Discussion
4.1. Atrial Remodeling in Patients with Active Colitis
4.2. Active Colitis-Induced Prolongation of Pdur and PR Interval
4.3. Colitis-Induced Changes in APD
4.4. The Gut-Heart Axis
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AF | Atrial fibrillation |
| Agt | Angiotensinogen |
| AngII | Angiotensin II |
| AP | Action potential |
| APD | Action potential duration |
| AT1R | Angiotensin II receptor type 1 |
| Cx | Connexin |
| DAI | Disease activity index |
| DSS | Dextran sulfate sodium |
| ECG | Electrocardiogram |
| IBD | Inflammatory bowel disease |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| LTCC | L-type Ca channel |
| Pdur | P wave duration |
| Pdisp | P wave dispersion |
| RAS | Renin-angiotensin system |
| SCFA | Short chain fatty acids |
| TMAO | Trimethylamine N-oxide |
| UC | Ulcerative colitis |
References
- Khan, M.W.; Keshavarzian, A.; Gounaris, E.; Melson, J.E.; Cheon, E.C.; Blatner, N.R.; Chen, Z.E.; Tsai, F.-N.; Lee, G.; Ryu, H.; et al. PI3K/AKT Signaling Is Essential for Communication between Tissue-Infiltrating Mast Cells, Macrophages, and Epithelial Cells in Colitis-Induced Cancer. Clin. Cancer Res. 2013, 19, 2342–2354. [Google Scholar] [CrossRef] [PubMed]
- Keerthivasan, S.; Aghajani, K.; Dose, M.; Molinero, L.; Khan, M.W.; Venkateswaran, V.; Weber, C.; Emmanuel, A.O.; Sun, T.; Bentrem, D.J.; et al. β-Catenin Promotes Colitis and Colon Cancer through Imprinting of Proinflammatory Properties in T Cells. Sci. Transl. Med. 2014, 6, 225ra28. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.; Navarini, A.; Rogler, G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, J.; Jess, T.; Kobylecki, C.J.; Nordestgaard, B.G.; Allin, K.H. Cardiovascular Risk Profile Among Patients with Inflammatory Bowel Disease: A Population-Based Study of More Than 100 000 Individuals. J. Crohns Colitis 2019, 13, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Bunu, D.-M.; Timofte, C.-E.; Ciocoiu, M.; Floria, M.; Tarniceriu, C.-C.; Barboi, O.-B.; Tanase, D.-M. Cardiovascular Manifestations of Inflammatory Bowel Disease: Pathogenesis, Diagnosis, and Preventive Strategies. Gastroenterol. Res. Pract. 2019, 2019, 3012509–3012514. [Google Scholar] [CrossRef]
- Mitchell, N.E.; Harrison, N.; Junga, Z.; Singla, M. Heart Under Attack: Cardiac Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 2322–2326. [Google Scholar] [CrossRef]
- Pattanshetty, D.J.; Anna, K.; Gajulapalli, R.D.; Shekkar, R.R.; Sappati-Biyyani, R. Inflammatory Bowel “Cardiac” Disease: Point Prevalence of Atrial Fibrillation in Inflammatory Bowel Disease Population. Saudi J. Gastroenterol. 2015, 21, 325–329. [Google Scholar] [CrossRef]
- Choi, Y.J.; Han, K.D.; Park, J.; Moon, I.; Lee, E.; Choe, W.S.; Lee, S.R.; Cha, M.J.; Lim, W.-H.; Oh, S. Increased Risk of Atrial Fibrillation in Patients with Inflammatory Bowel Disease: A Nationwide Population-Based Study. World J. Gastroenterol. 2019, 25, 2788–2798. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Kristensen, S.L.; Lindhardsen, J.; Lindhardsen, J.; Ahlehoff, O.; Ahlenhoff, O.; Erichsen, R.; Erichsen, R.; Lamberts, M.; Lamberts, M.; et al. Increased Risk of Atrial Fibrillation and Stroke during Active Stages of Inflammatory Bowel Disease: A Nationwide Study. Europace 2014, 16, 477–484. [Google Scholar] [CrossRef]
- Sun, J.; Roelstraete, B.; Svennberg, E.; Halfvarson, J.; Sundström, J.; Forss, A.; Olén, O.; Ludvigsson, J.F. Long-Term Risk of Arrhythmias in Patients with Inflammatory Bowel Disease: A Population-Based, Sibling-Controlled Cohort Study. PLoS Med. 2023, 20, e1004305. [Google Scholar] [CrossRef]
- Bornaun, H.A.; Yılmaz, N.; Kutluk, G.; Dedeoğlu, R.; Oztarhan, K.; Keskindemirci, G.; Tulunoğlu, A.; Şap, F. Prolonged P-Wave and QT Dispersion in Children with Inflammatory Bowel Disease in Remission. BioMed Res. Int. 2017, 2017, 6960810. [Google Scholar] [CrossRef] [PubMed]
- Curione, M.; Aratari, A.; Amato, S.; Colotto, M.; Barbato, M.; Leone, S.; Tego, A.; Panetti, D.; Parlapiano, C. A Study on QT Interval in Patients Affected with Inflammatory Bowel Disease without Cardiac Involvement. Intern. Emerg. Med. 2010, 5, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Efe, T.H.; Cimen, T.; Ertem, A.G.; Coskun, Y.; Bilgin, M.; Sahan, H.F.; Pamukcu, H.E.; Yayla, C.; Sunman, H.; Yuksel, I.; et al. Atrial Electromechanical Properties in Inflammatory Bowel Disease. Echocardiography 2016, 33, 1309–1316. [Google Scholar] [CrossRef]
- Nar, G.; Ergul, B.; Aksan, G.; Inci, S. Assessment of Atrial Electromechanical Delay and Left Atrial Mechanical Functions in Patients with Ulcerative Colitis. Echocardiography 2016, 33, 970–976. [Google Scholar] [CrossRef]
- Rasmussen, M.U.; Kumarathurai, P.; Fabricius-Bjerre, A.; Larsen, B.S.; Domínguez, H.; Davidsen, U.; Gerds, T.A.; Kanters, J.K.; Sajadieh, A. P-wave Indices as Predictors of Atrial Fibrillation. Ann. Noninvasive Electrocardiol. 2020, 25, e12751. [Google Scholar] [CrossRef] [PubMed]
- Baturova, M.A.; Cornefjord, G.; Carlson, J.; Johnson, L.S.B.; Smith, J.G.; Platonov, P.G. P-Wave Characteristics as Electrocardiographic Markers of Atrial Abnormality in Prediction of Incident Atrial Fibrillation—The Malmö Preventive Project. J. Electrocardiol. 2024, 82, 125–130. [Google Scholar] [CrossRef]
- Giannopoulos, G.; Tachmatzidis, D.; Moysidis, D.V.; Filos, D.; Petridou, M.; Chouvarda, I.; Vassilikos, V.P. P-Wave Indices as Predictors of Atrial Fibrillation: The Lion from a Claw. Curr. Probl. Cardiol. 2024, 49, 102051. [Google Scholar] [CrossRef]
- Censi, F.; Corazza, I.; Reggiani, E.; Calcagnini, G.; Mattei, E.; Triventi, M.; Boriani, G. P-Wave Variability and Atrial Fibrillation. Sci. Rep. 2016, 6, 26799. [Google Scholar] [CrossRef]
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Belke, D.D.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. Distinct Patterns of Atrial Electrical and Structural Remodeling in Angiotensin II Mediated Atrial Fibrillation. J. Mol. Cell. Cardiol. 2018, 124, 12–25. [Google Scholar] [CrossRef]
- Rohr, S. Role of Gap Junctions in the Propagation of the Cardiac Action Potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Jeevaratnam, K.; Guzadhur, L.; Goh, Y.M.; Grace, A.A.; Huang, C.L.-H. Sodium Channel Haploinsufficiency and Structural Change in Ventricular Arrhythmogenesis. Acta Physiol. 2016, 216, 186–202. [Google Scholar] [CrossRef]
- Bao, Y.; Willis, B.C.; Frasier, C.R.; Lopez-Santiago, L.F.; Lin, X.; Ramos-Mondragón, R.; Auerbach, D.S.; Chen, C.; Wang, Z.; Anumonwo, J.; et al. Scn2b Deletion in Mice Results in Ventricular and Atrial Arrhythmias. Circ. Arrhythmia Electrophysiol. 2018, 9, e003923. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium Channel Mutations and Susceptibility to Heart Failure and Atrial Fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef]
- Chen, L.; Ballew, J.; Herron, K.; Rodeheffer, R.; Olson, T. A Common Polymorphism in SCN5A Is Associated with Lone Atrial Fibrillation. Clin. Pharmacol. Ther. 2007, 81, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh-Isleem, A.; Clatot, J.; Duchatelet, S.; Gandjbakhch, E.; Denjoy, I.; Hidden-Lucet, F.; Hatem, S.; Deschênes, I.; Coulombe, A.; Neyroud, N.; et al. A Truncating SCN5A Mutation Combined with Genetic Variability Causes Sick Sinus Syndrome and Early Atrial Fibrillation. Heart Rhythm 2014, 11, 1015–1023. [Google Scholar] [CrossRef]
- Gawałko, M.; Agbaedeng, T.A.; Saljic, A.; Müller, D.N.; Wilck, N.; Schnabel, R.; Penders, J.; Rienstra, M.; van Gelder, I.; Jespersen, T.; et al. Gut Microbiota, Dysbiosis and Atrial Fibrillation. Arrhythmogenic Mechanisms and Potential Clinical Implications. Cardiovasc. Res. 2021, 118, 2415–2427. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Jackson, K.L.; Gueguen, C.; Lim, K.; Eikelis, N.; Stevenson, E.R.; Charchar, F.J.; Lambert, G.W.; Burke, S.L.; Paterson, M.R.; Marques, F.Z.; et al. Neural Suppression of MiRNA-181a in the Kidney Elevates Renin Expression and Exacerbates Hypertension in Schlager Mice. Hypertens. Res. 2020, 43, 1152–1164. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef]
- Liu, W.; Tan, Z.; Geng, M.; Jiang, X.; Xin, Y. Impact of the Gut Microbiota on Angiotensin II-Related Disorders and Its Mechanisms. Biochem. Pharmacol. 2023, 214, 115659. [Google Scholar] [CrossRef]
- Pereira, C.H.; Kittaka, H.; Ouille V, E.J.; Almeida, J.F.Q.; Pelaez, A.; Keshavarzian, A.; Blatter, L.A.; Banach, K. Colitis Induced Ventricular Alternans Increases the Risk for Ventricular Arrhythmia. J. Mol. Cell. Cardiol. 2025, 204, 68–78. [Google Scholar] [CrossRef]
- Goette, A.; Staack, T.; Röcken, C.; Arndt, M.; Geller, J.C.; Huth, C.; Ansorge, S.; Klein, H.U.; Lendeckel, U. Increased Expression of Extracellular Signal-Regulated Kinase and Angiotensin-Converting Enzyme in Human Atria during Atrial Fibrillation. J. Am. Coll. Cardiol. 2000, 35, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Li, D.; Thorin-Trescases, N.; Leung, T.-K.; Thorin, E.; Nattel, S. Evolution of the Atrial Fibrillation Substrate in Experimental Congestive Heart Failure: Angiotensin-Dependent and -Independent Pathways. Cardiovasc. Res. 2003, 60, 315–325. [Google Scholar] [CrossRef]
- Desantiago, J.; Bare, D.J.; Varma, D.; Solaro, R.J.; Arora, R.; Banach, K. Loss of P21-Activated Kinase 1 (Pak1) Promotes Atrial Arrhythmic Activity. Heart Rhythm 2018, 15, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Perše, M.; Perse, M.; Cerar, A.; Cerar, A. Dextran Sodium Sulphate Colitis Mouse Model: Traps and Tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.F.; Leite, D.F.P.; Marcon, R.; Claudino, R.F.; Dutra, R.C.; Cola, M.; Martini, A.C.; Calixto, J.B. Evaluation of Chemical Mediators and Cellular Response during Acute and Chronic Gut Inflammatory Response Induced by Dextran Sodium Sulfate in Mice. Biochem. Pharmacol. 2012, 84, 1459–1469. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Lu, H.; Yuan, P.; Ma, X.; Jiang, X.; Liu, S.; Ma, C.; Philipsen, S.; Zhang, Q.; Yang, J.; Xu, F.; et al. Angiotensin-Converting Enzyme Inhibitor Promotes Angiogenesis through Sp1/Sp3-Mediated Inhibition of Notch Signaling in Male Mice. Nat. Commun. 2023, 14, 731. [Google Scholar] [CrossRef]
- Pereira, C.H.; Bare, D.J.; Rosas, P.C.; Dias, F.A.L.; Banach, K. The Role of P21-Activated Kinase (Pak1) in Sinus Node Function. J. Mol. Cell. Cardiol. 2023, 179, 90–101. [Google Scholar] [CrossRef]
- Desantiago, J.; Bare, D.J.; Ke, Y.; Sheehan, K.A.; Solaro, R.J.; Banach, K. Functional Integrity of the T-Tubular System in Cardiomyocytes Depends on P21-Activated Kinase. J. Mol. Cell. Cardiol. 2013, 60, 121–128. [Google Scholar] [CrossRef]
- Varma, D.; Almeida, J.F.Q.; DeSantiago, J.; Blatter, L.A.; Banach, K. Inositol 1,4,5-Trisphosphate Receptor-Reactive Oxygen Signaling Domain Regulates Excitation-Contraction Coupling in Atrial Myocytes. J. Mol. Cell. Cardiol. 2022, 163, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.; Egert, U.; Hescheler, J.; Banach, K. Estimation of Action Potential Changes from Field Potential Recordings in Multicellular Mouse Cardiac Myocyte Cultures. Cell. Physiol. Biochem. 2003, 13, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, S.; Kim, J.S.; Hagendorff, A.; Thonnissen, E.; Kruger, O.; Lamers, W.H.; Willecke, K. Abnormal Cardiac Conduction and Morphogenesis in Connexin40 and Connexin43 Double-Deficient Mice. Circ. Res. 2000, 87, 399. [Google Scholar] [CrossRef]
- Kanagaratnam, P.; Peters, N.S. Conduction, Gap Junctions, and Atrial Fibrillation: An Eternal Triangle? Heart Rhythm 2004, 1, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Remme, C.A.; Scicluna, B.P.; Verkerk, A.O.; Amin, A.S.; van Brunschot, S.; Beekman, L.; Deneer, V.H.M.; Chevalier, C.; Oyama, F.; Miyazaki, H.; et al. Genetically Determined Differences in Sodium Current Characteristics Modulate Conduction Disease Severity in Mice with Cardiac Sodium Channelopathy. Circ. Res. 2009, 104, 1283–1292. [Google Scholar] [CrossRef]
- Remme, C.A. SCN5A Channelopathy: Arrhythmia, Cardiomyopathy, Epilepsy and Beyond. Philos. Trans. R. Soc. B 2023, 378, 20220164. [Google Scholar] [CrossRef]
- Mercier, A.; Clément, R.; Harnois, T.; Bourmeyster, N.; Bois, P.; Chatelier, A. Nav1.5 Channels Can Reach the Plasma Membrane through Distinct N-Glycosylation States. Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 1215–1223. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Darbar, D. Atrial Fibrillation and SCN5A Variants. Card. Electrophysiol. Clin. 2014, 6, 741–748. [Google Scholar] [CrossRef]
- Regan, C.P.; Wallace, A.A.; Cresswell, H.K.; Atkins, C.L.; Lynch, J.J. In Vivo Cardiac Electrophysiologic Effects of a Novel Diphenylphosphine Oxide IKur Blocker, (2-Isopropyl-5-Methylcyclohexyl) Diphenylphosphine Oxide, in Rat and Nonhuman Primate. J. Pharmacol. Exp. Ther. 2006, 316, 727–732. [Google Scholar] [CrossRef]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.-E.; Conklin, L.S.; Centola, M.; Li, X. Distinct Cytokine Patterns Identified from Multiplex Profiles of Murine DSS and TNBS-Induced Colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef]
- De Oliveira, Í.M.; da Silva Júnior, E.L.; Martins, Y.D.O.; Rocha, H.A.L.; Scanavacca, M.I.; Gutierrez, P.S. Cardiac Autonomic Nervous System Remodeling May Play a Role in Atrial Fibrillation: A Study of the Autonomic Nervous System and Myocardial Receptors. Arq. Bras. Cardiol. 2021, 117, 999–1007. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Dobrev, D.; Nattel, S. Atrial Fibrillation: Pathophysiology, Genetic and Epigenetic Mechanisms. Lancet Reg. Health Eur. 2024, 37, 100785. [Google Scholar] [CrossRef]
- Kang, W.; Deng, J.; Fan, Z.; Zhou, F.; Wang, X.; Liu, K.; Wang, L. Mechanisms Associated with the Development of Atrial Fibrillation after Sepsis and the Role of Neuregulin-1. Preprint 2023. [Google Scholar] [CrossRef]
- Joukar, S. A Comparative Review on Heart Ion Channels, Action Potentials and Electrocardiogram in Rodents and Human: Extrapolation of Experimental Insights to Clinic. Lab. Anim. Res. 2021, 37, 25. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Argenziano, M.; Antzelevitch, C. Cardiac Arrhythmias Related to Sodium Channel Dysfunction. Handb. Exp. Pharmacol. 2017, 246, 331–354. [Google Scholar] [CrossRef]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and Molecular Electrophysiology of Atrial Fibrillation Initiation, Maintenance, and Progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.J.M.; Gelder, I.C.V.; Henning, R.H.; Tuinenburg, A.E.; Wietses, M.; Grandjean, J.G.; Wilde, A.A.M.; Gilst, W.H.V.; Crijns, H.J.G.M. Alterations in Potassium Channel Gene Expression in Atria of Patients with Persistent and Paroxysmal Atrial Fibrillation: Differential Regulation of Protein and MRNA Levels for K+ Channels. J. Am. Coll. Cardiol. 2001, 37, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Nichols, C.G.; Schwarz, T.L.; Escande, D. Genetic Manipulation of Cardiac K+ Channel Function in Mice. Circ. Res. 2001, 89, 944–956. [Google Scholar] [CrossRef]
- Wagoner, D.R.V.; Pond, A.L.; McCarthy, P.M.; Trimmer, J.S.; Nerbonne, J.M. Outward K+ Current Densities and Kv1.5 Expression Are Reduced in Chronic Human Atrial Fibrillation. Circ. Res. 1997, 80, 772–781. [Google Scholar] [CrossRef]
- Bosch, R.F.; Scherer, C.R.; Rüb, N.; Wöhrl, S.; Steinmeyer, K.; Haase, H.; Busch, A.E.; Seipel, L.; Kühlkamp, V. Molecular Mechanisms of Early Electrical Remodeling: Transcriptional Downregulation of Ion Channel Subunits Reduces ICa,L and Ito in Rapid Atrial Pacing in Rabbits. J. Am. Coll. Cardiol. 2003, 41, 858–869. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.; Lin, X.; Yang, Y.; Hong, K.; Wang, L.; Liu, J.; Li, L.; Yan, D.; Liang, D.; et al. Novel KCNA5 Loss-of-Function Mutations Responsible for Atrial Fibrillation. J. Hum. Genet. 2009, 54, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular Physiology of Cardiac Repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Li, H.; London, B.; Nerbonne, J.M. Functional Consequences of Elimination of I(to,f) and I(to,s): Early Afterdepolarizations, Atrioventricular Block, and Ventricular Arrhythmias in Mice Lacking Kv1.4 and Expressing a Dominant-Negative Kv4 Alpha Subunit. Circ. Res. 2000, 87, 73–79. [Google Scholar] [CrossRef]
- Xu, H.; Li, H.; Nerbonne, J.M. Elimination of the Transient Outward Current and Action Potential Prolongation in Mouse Atrial Myocytes Expressing a Dominant Negative Kv4 α Subunit. J. Physiol. 1999, 519, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ordás, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative Colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef]
- Sinha, T.; Zain, Z.; Bokhari, S.F.H.; Waheed, S.; Reza, T.; Eze-Odurukwe, A.; Patel, M.; Almadhoun, M.K.I.K.; Hussain, A.; Reyaz, I. Navigating the Gut-Cardiac Axis: Understanding Cardiovascular Complications in Inflammatory Bowel Disease. Cureus 2024, 16, e55268. [Google Scholar] [CrossRef]
- Yu, L.; Meng, G.; Huang, B.; Zhou, X.; Stavrakis, S.; Wang, M.; Li, X.; Zhou, L.; Wang, Y.; Wang, M.; et al. A Potential Relationship between Gut Microbes and Atrial Fibrillation: Trimethylamine N-Oxide, a Gut Microbe-Derived Metabolite, Facilitates the Progression of Atrial Fibrillation. Int. J. Cardiol. 2018, 255, 92–98. [Google Scholar] [CrossRef]
- Jaworska, K.; Koper, M.; Ufnal, M. Gut Microbiota and Renin-Angiotensin System: A Complex Interplay at Local and Systemic Levels. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 321, G355–G366. [Google Scholar] [CrossRef]
- Salmenkari, H.; Korpela, R.; Vapaatalo, H. Renin–Angiotensin System in Intestinal Inflammation—Angiotensin Inhibitors to Treat Inflammatory Bowel Diseases? Basic Clin. Pharmacol. Toxicol. 2021, 129, 161–172. [Google Scholar] [CrossRef]
- Higham, J.P.; Bhebhe, C.N.; Gupta, R.A.; Tranter, M.M.; Barakat, F.M.; Dogra, H.; Bab, N.; Wozniak, E.; Barker, K.H.; Wilson, C.H.; et al. Transcriptomic Profiling Reveals a Pronociceptive Role for Angiotensin II in Inflammatory Bowel Disease. Pain 2024, 165, 1592–1604. [Google Scholar] [CrossRef]
- Nowak, J.K.; Lindstrøm, J.C.; Kalla, R.; Ricanek, P.; Halfvarson, J.; Satsangi, J. Age, Inflammation, and Disease Location Are Critical Determinants of Intestinal Expression of SARS-CoV-2 Receptor ACE2 and TMPRSS2 in Inflammatory Bowel Disease. Gastroenterology 2020, 159, 1151–1154.e2. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.U.; Yang, H.; Haxhija, E.Q.; Wildhaber, B.E.; Greenson, J.K.; Teitelbaum, D.H. Reduced Severity of a Mouse Colitis Model with Angiotensin Converting Enzyme Inhibition. Dig. Dis. Sci. 2007, 52, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Katada, K.; Yoshida, N.; Suzuki, T.; Okuda, T.; Mizushima, K.; Takagi, T.; Ichikawa, H.; Naito, Y.; Cepinskas, G.; Yoshikawa, T. Dextran Sulfate Sodium-Induced Acute Colonic Inflammation in Angiotensin II Type 1a Receptor Deficient Mice. Inflamm. Res. 2008, 57, 84–91. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Borges, J.I.; Stoicovy, R.A. RGS Proteins and Cardiovascular Angiotensin II Signaling: Novel Opportunities for Therapeutic Targeting. Biochem. Pharmacol. 2023, 218, 115904. [Google Scholar] [CrossRef] [PubMed]
- Demers, J.; Ton, A.; Huynh, F.; Thibault, S.; Ducharme, A.; Paradis, P.; Nemer, M.; Fiset, C. Atrial Electrical Remodeling in Mice with Cardiac-Specific Overexpression of Angiotensin II Type 1 Receptor. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2021, 11, e023974. [Google Scholar] [CrossRef]
- Gu, J.; Hu, W.; Liu, X. Pioglitazone Improves Potassium Channel Remodeling Induced by Angiotensin II in Atrial Myocytes. Med. Sci. Monit. Basic Res. 2014, 20, 153–160. [Google Scholar] [CrossRef]
- Oh, S.Y.; Cho, K.-A.; Kang, J.L.; Kim, K.H.; Woo, S.-Y. Comparison of Experimental Mouse Models of Inflammatory Bowel Disease. Int. J. Mol. Med. 2014, 33, 333–340. [Google Scholar] [CrossRef]
- Chen, L.; Fu, G.; Jiang, C. Mendelian Randomization as an Approach to Assess Causal Effects of Inflammatory Bowel Disease on Atrial Fibrillation. Aging 2021, 13, 12016–12030. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTL | DSSA | DSSR | p Values | ||||
|---|---|---|---|---|---|---|---|
| Activation | n | 37/6 | 31/6 | 20/2 | CTL vs. DSSA | CTL vs. DSSR | DSSA vs. DSSR |
| V1/2a | −53.3 ± 0.2 | −49.2 ± 0.2 | −50.1 ± 0.2 | <0.001 | <0.001 | 0.025 | |
| slope | 2.5 ± 0.2 | 3.0 ± 0.2 | 2.6 ± 0.2 | 0.096 | 0.932 | 0.327 | |
| Inactivation | n | 37/6 | 31/6 | 20/2 | |||
| V1/2i | −79.2 ± 0.4 | −83.3 ± 0.3 | −80.7 ± 0.5 | <0.001 | 0.024 | <0.001 | |
| slope | −8.7 ± 0.3 | −9.1 ± 0.3 | −8.2 ± 0.4 | 0.535 | 0.634 | 0.179 | |
| CTL | DSSA | DSSi(ACE) | p Values | ||||
|---|---|---|---|---|---|---|---|
| Activation | n | 37/6 | 31/6 | 32/6 | CTL vs. DSSA | CTL vs. DSSi(ACE) | DSSA vs. DSSi(ACE) |
| V1/2a | −53.3 ± 0.2 | −49.2 ± 0.2 | −49.0 ± 0.3 | <0.001 | <0.001 | 0.827 | |
| slope | 2.5 ± 0.2 | 3.0 ± 0.2 | 3.8 ± 0.2 | 0.162 | <0.001 | 0.006 | |
| Inactivation | n | 37/6 | 31/6 | 32/6 | |||
| V1/2i | −79.2 ± 0.4 | −83.3 ± 0.3 | −85.8 ± 0.4 | <0.001 | <0.001 | <0.001 | |
| slope | −8.7 ± 0.3 | −9.1 ± 0.3 | −8.9 ± 0.4 | 0.727 | 0.997 | 0.785 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kittaka, H.; Ouille V, E.J.; Pereira, C.H.; Pélaez, A.F.; Keshavarzian, A.; Banach, K. Active Colitis-Induced Atrial Electrophysiological Remodeling. Biomolecules 2025, 15, 982. https://doi.org/10.3390/biom15070982
Kittaka H, Ouille V EJ, Pereira CH, Pélaez AF, Keshavarzian A, Banach K. Active Colitis-Induced Atrial Electrophysiological Remodeling. Biomolecules. 2025; 15(7):982. https://doi.org/10.3390/biom15070982
Chicago/Turabian StyleKittaka, Hiroki, Edward J. Ouille V, Carlos H. Pereira, Andrès F. Pélaez, Ali Keshavarzian, and Kathrin Banach. 2025. "Active Colitis-Induced Atrial Electrophysiological Remodeling" Biomolecules 15, no. 7: 982. https://doi.org/10.3390/biom15070982
APA StyleKittaka, H., Ouille V, E. J., Pereira, C. H., Pélaez, A. F., Keshavarzian, A., & Banach, K. (2025). Active Colitis-Induced Atrial Electrophysiological Remodeling. Biomolecules, 15(7), 982. https://doi.org/10.3390/biom15070982