
The Proteomic Landscape of Parkin-Deficient and Parkin-Overexpressing Rat Nucleus Accumbens: An Insight into the Role of Parkin in Methamphetamine Use Disorder

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
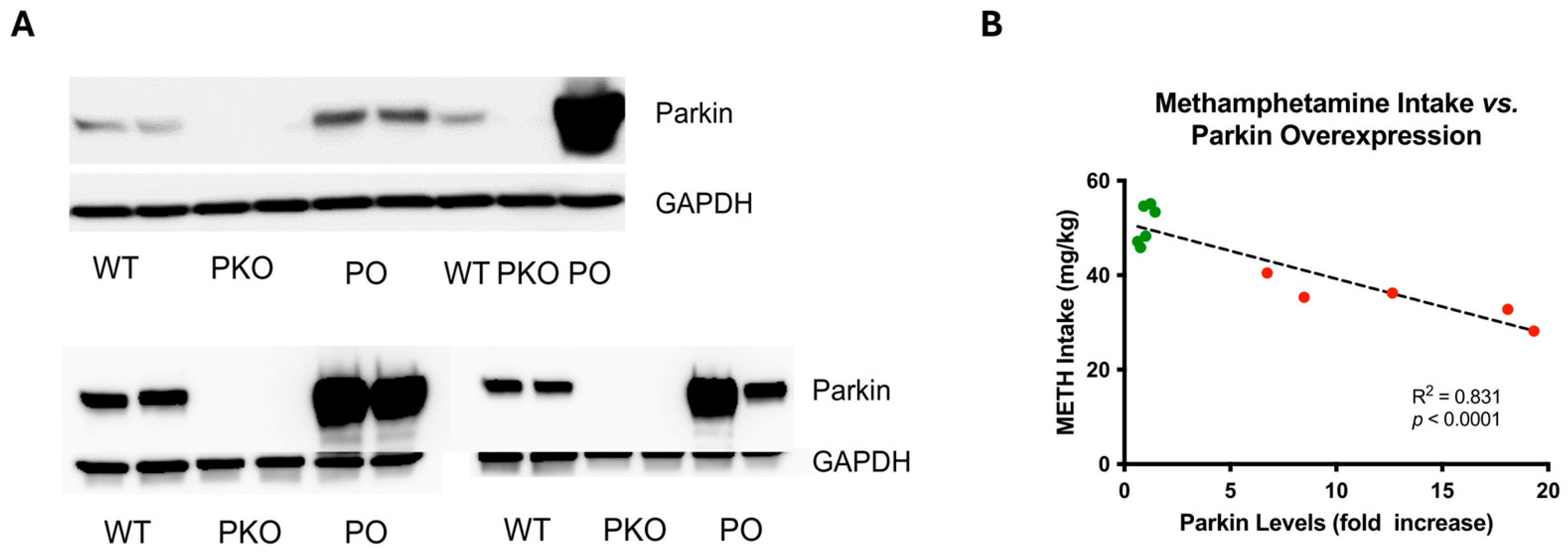
2.2. Parkin Overexpression Employing the Adeno-Associated Viral Transfer Vector
2.3. Tissue Collection and Sample Preparation
2.3.1. LC-MS/MS
2.3.2. SDS-PAGE and Western Blotting
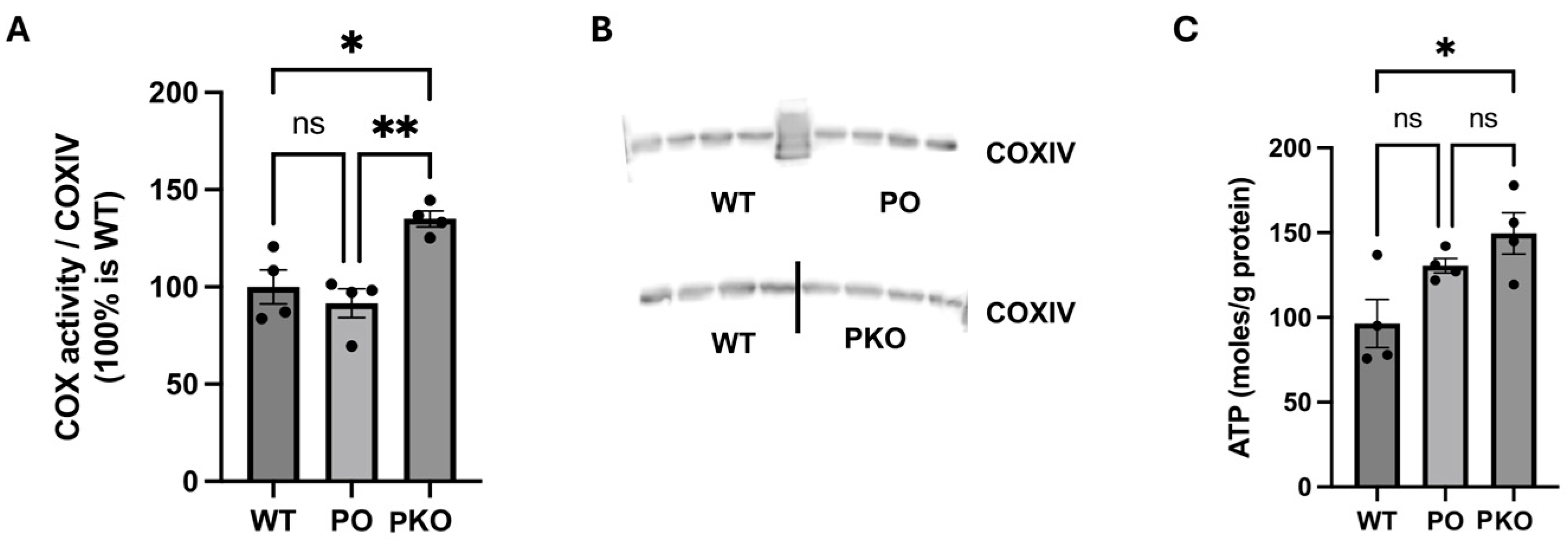
2.3.3. Complex IV Activity and ATP Levels
2.4. Label-Free Quantification (LFQ) Data Collection
2.5. SDS-PAGE and Western Blotting
2.6. Complex IV-Specific Activity and ATP Levels
2.7. Data Analyses
2.7.1. LFQ Data Analysis
2.7.2. Molecular Biology Data Analysis
3. Results
3.1. Differentially Expressed Proteins in Rat Nucleus Accumbens Lacking Parkin
3.2. Differentially Expressed Proteins in Rat Nucleus Accumbens Overexpressing Parkin
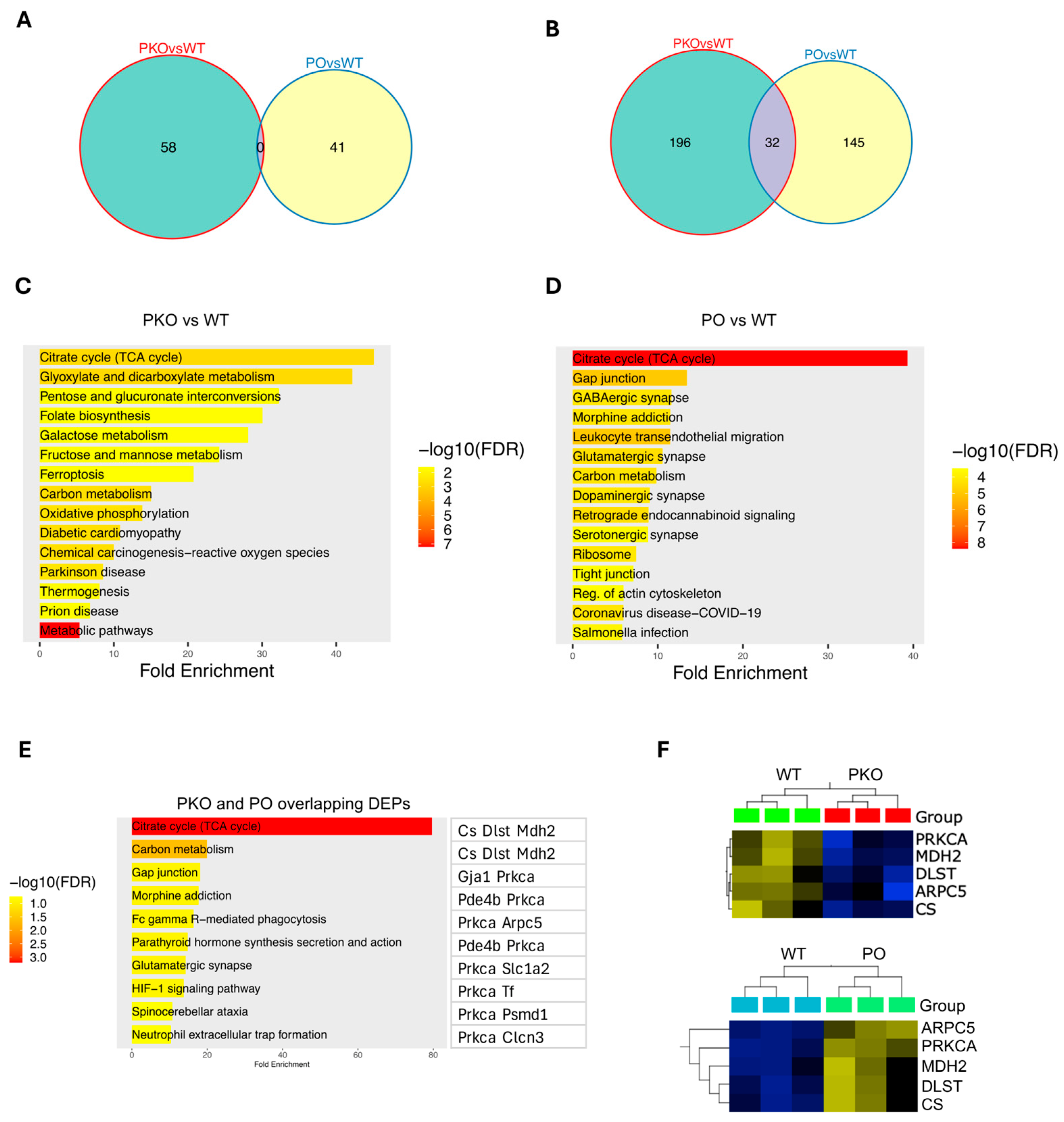
3.3. Molecular Pathways Changed by Parkin Knockout or Parkin Overexpression in Rat Nucleus Accumbens
3.4. Molecular Pathways Changed by Parkin Knockout and Parkin Overexpression in Rat Nucleus Accumbens
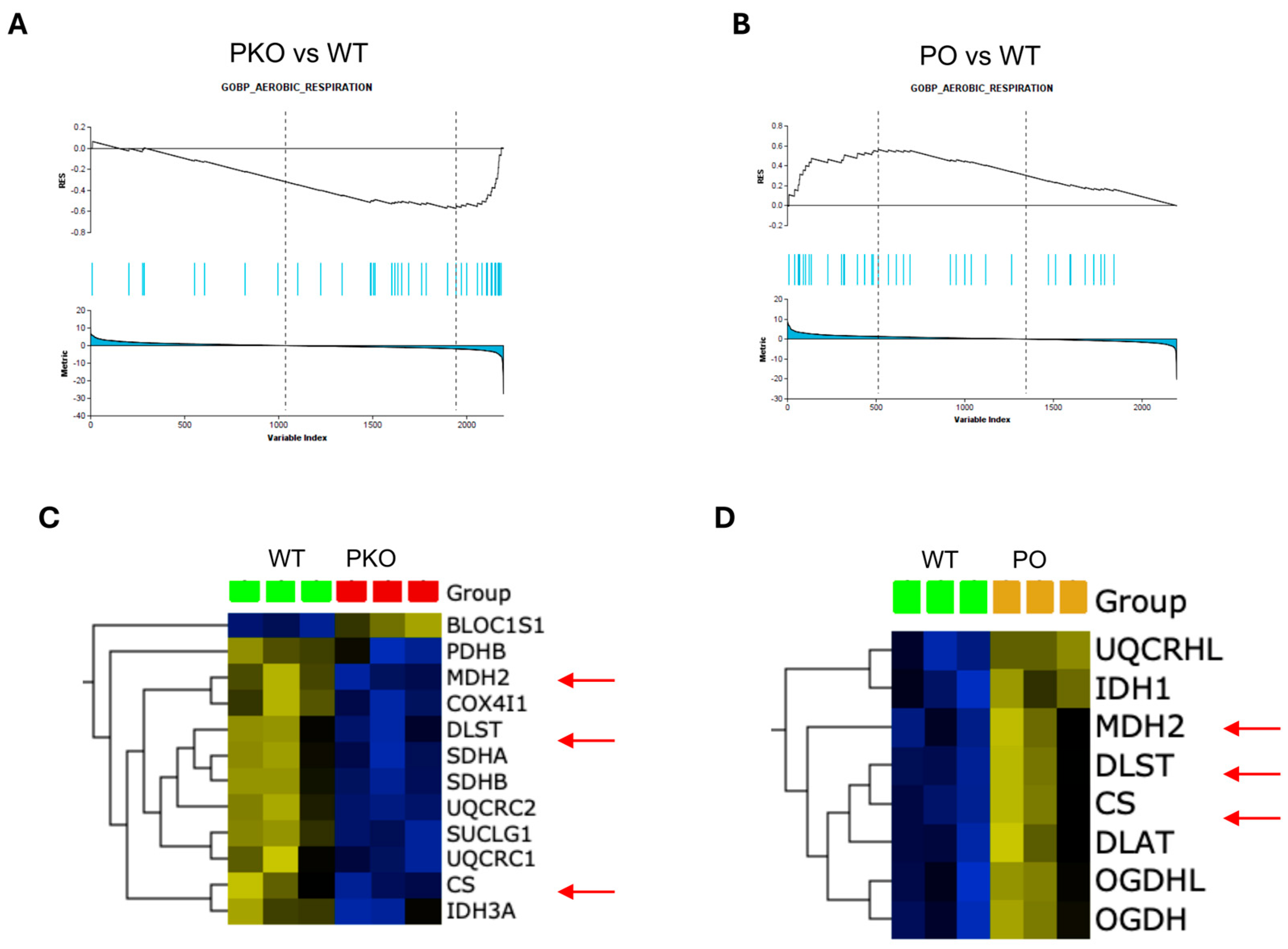
3.5. Mitochondrial Function in Parkin-Deficient and Parkin-Overexpressing Nucleus Accumbens
3.6. Genes Coding for Differentially Expressed Proteins in Parkin-Deficient and Parkin-Overexpressing Nucleus Accumbens Overlap with Genes Associated with MUD
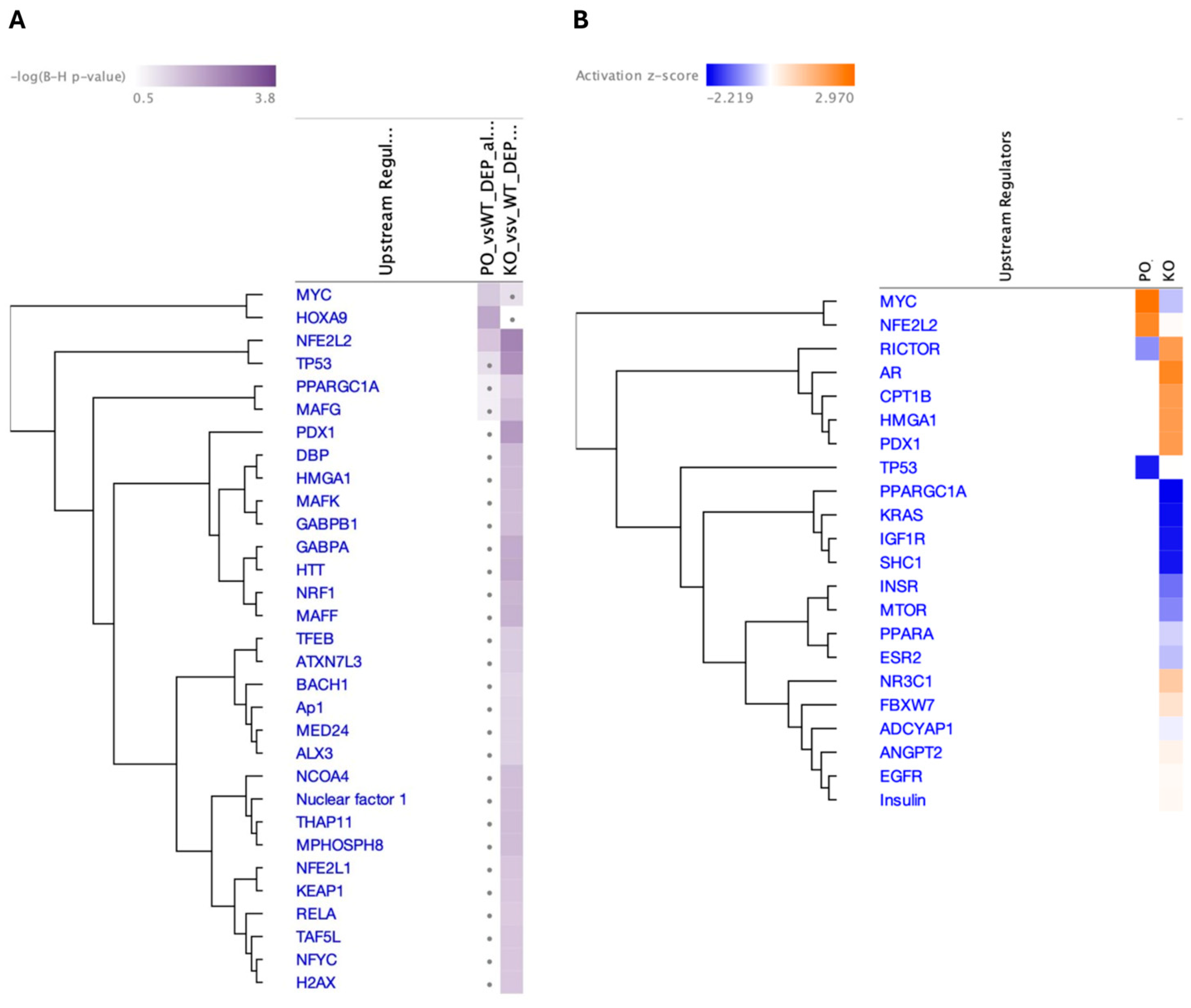
3.7. Top Upstream Transcriptional Regulators Prediction by IPA
4. Discussion
4.1. Parkin and Energy Metabolism in Rat Nucleus Accumbens
4.2. Parkin and Neurotransmission in Rat Nucleus Accumbens
4.3. Parkin and Actin Cytoskeleton Dynamics in Rat Nucleus Accumbens
4.4. Parkin and Differentially Expressed Proteins
4.5. Parkin and Genetics of Methamphetamine Use Disorder
5. Conclusions
6. Limitations and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jalal, H.; Buchanich, J.M.; Roberts, M.S.; Balmert, L.C.; Zhang, K.; Burke, D.S. Changing dynamics of the drug overdose epidemic in the United States from 1979 through 2016. Science 2018, 361, eaau1184. [Google Scholar] [CrossRef]
- NIDA. Overdose Death Rates; NIDA: North Bethesda, MD, USA, 2023. [Google Scholar]
- Guerin, A.A.; Nestler, E.J.; Berk, M.; Lawrence, A.J.; Rossell, S.L.; Kim, J.H. Genetics of methamphetamine use disorder: A systematic review and meta-analyses of gene association studies. Neurosci. Biobehav. Rev. 2021, 120, 48–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Harutyunyan, A.; Schneider, B.L.; Moszczynska, A. Parkin regulates drug-taking behavior in rat model of methamphetamine use disorder. Transl. Psych. 2021, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J. Parkin: A multifaceted ubiquitin ligase. Biochem. Soc. Trans. 2006, 34 Pt 5, 749–753. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Zhao, J.; Feng, J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum. Mol. Genet. 2004, 13, 1745–1754. [Google Scholar] [CrossRef]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmun Pharmacol. 2014, 9, 569–581. [Google Scholar] [CrossRef]
- Schmidt, S.; Linnartz, B.; Mendritzki, S.; Sczepan, T.; Lubbert, M.; Stichel, C.C.; Lubbert, H. Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum. Mol. Gen. 2011, 20, 1197–1211. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, Q.; Feng, J. Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J. Biol. Chem. 2004, 279, 54380–54386. [Google Scholar] [CrossRef]
- Hyun, D.H.; Lee, M.; Hattori, N.; Kubo, S.; Mizuno, Y.; Halliwell, B.; Jenner, P. Effect of wild-type or mutant Parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J. Biol. Chem. 2002, 277, 28572–28577. [Google Scholar] [CrossRef]
- Sassone, J.; Serratto, G.; Valtorta, F.; Silani, V.; Passafaro, M.; Ciammola, A. The synaptic function of parkin. Brain 2017, 140, 2265–2272. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- Jiang, H.; Ren, Y.; Yuen, E.Y.; Zhong, P.; Ghaedi, M.; Hu, Z.; Azabdaftari, G.; Nakaso, K.; Yan, Z.; Feng, J. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 2012, 3, 668. [Google Scholar] [CrossRef] [PubMed]
- Moszczynska, A.; Saleh, J.; Zhang, H.; Vukusic, B.; Lee, F.J.; Liu, F. Parkin disrupts the alpha-synuclein/dopamine transporter interaction: Consequences toward dopamine-induced toxicity. J. Mol. Neurosci. 2007, 32, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.P.; Scoles, D.R.; Ho, T.H.; Del Bigio, M.R.; Pulst, S.M. Parkin is associated with actin filaments in neuronal and nonneural cells. Ann. Neurol. 2000, 48, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, J.; Feng, J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J. Neurosci. 2003, 23, 3316–3324. [Google Scholar] [CrossRef]
- Martella, G.; Platania, P.; Vita, D.; Sciamanna, G.; Cuomo, D.; Tassone, A.; Tscherter, A.; Kitada, T.; Bonsi, P.; Shen, J.; et al. Enhanced sensitivity to group II mGlu receptor activation at corticostriatal synapses in mice lacking the familial parkinsonism-linked genes PINK1 or Parkin. Exp. Neurol. 2009, 215, 388–396. [Google Scholar] [CrossRef]
- Jang, E.Y.; Yang, C.H.; Hedges, D.M.; Kim, S.P.; Lee, J.Y.; Ekins, T.G.; Garcia, B.T.; Kim, H.Y.; Nelson, A.C.; Kim, N.J.; et al. The role of reactive oxygen species in methamphetamine self-administration and dopamine release in the nucleus accumbens. Addict. Biol. 2017, 22, 1304–1315. [Google Scholar] [CrossRef]
- Najera, J.A.; Bustamante, E.A.; Bortell, N.; Morsey, B.; Fox, H.S.; Ravasi, T.; Marcondes, M.C. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016, 17, 7. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Raudensky, J. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J. Neuroimmune Pharmacol. 2008, 3, 203–217. [Google Scholar] [CrossRef]
- Stephans, S.E.; Whittingham, T.S.; Douglas, A.J.; Lust, W.D.; Yamamoto, B.K. Substrates of energy metabolism attenuate methamphetamine-induced neurotoxicity in striatum. J. Neurochem. 1998, 71, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Volkow, N.D.; Chang, L.; Miller, E.; Sedler, M.; Hitzemann, R.; Zhu, W.; Logan, J.; Ma, Y.; Fowler, J.S. Partial recovery of brain metabolism in methamphetamine abusers after protracted abstinence. Am. J. Psychiatry 2004, 161, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Smith, L.; Fowler, J.S.; Telang, F.; Logan, J.; Tomasi, D. Recovery of dopamine transporters with methamphetamine detoxification is not linked to changes in dopamine release. NeuroImage 2015, 121, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Ouchi, Y.; Sugihara, G.; Takei, N.; Yoshikawa, E.; Nakamura, K.; Iwata, Y.; Tsuchiya, K.J.; Suda, S.; Suzuki, K.; et al. Methamphetamine causes microglial activation in the brains of human abusers. J. Neurosc. 2008, 28, 5756–5761. [Google Scholar] [CrossRef]
- Young, E.J.; Briggs, S.B.; Miller, C.A. The Actin Cytoskeleton as a Therapeutic Target for the Prevention of Relapse to Methamphetamine Use. CNS Neurol. Disord. Drug Targets 2015, 14, 731–737. [Google Scholar] [CrossRef]
- Moussawi, K.; Kalivas, P.W. Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur. J. Pharmacol. 2010, 639, 115–122. [Google Scholar] [CrossRef]
- Jayanthi, S.; McCoy, M.T.; Chen, B.; Britt, J.P.; Kourrich, S.; Yau, H.J.; Ladenheim, B.; Krasnova, I.N.; Bonci, A.; Cadet, J.L. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol. Psych. 2014, 76, 47–56. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Justinova, Z.; Cadet, J.L. Methamphetamine addiction: Involvement of CREB and neuroinflammatory signaling pathways. Psychopharmacology 2016, 233, 1945–1962. [Google Scholar] [CrossRef]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.; Roberts-Wolfe, D.; Kalivas, P.W. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar]
- Dusonchet, J.; Bensadoun, J.C.; Schneider, B.L.; Aebischer, P. Targeted overexpression of the parkin substrate Pael-R in the nigrostriatal system of adult rats to model Parkinson’s disease. Neurobiol. Dis. 2009, 35, 32–41. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Lee, I.; Salomon, A.R.; Ficarro, S.; Mathes, I.; Lottspeich, F.; Grossman, L.I.; Huttemann, M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J. Biol. Chem. 2005, 280, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Huttemann, M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef] [PubMed]
- Alves da Costa, C.; Duplan, E.; Rouland, L.; Checler, F. The Transcription Factor Function of Parkin: Breaking the Dogma. Front. Neurosci. 2018, 12, 965. [Google Scholar] [CrossRef]
- Crowder, T.L.; Weiner, J.L. Functional characterization of kainate receptors in the rat nucleus accumbens core region. J. Neurophysiol. 2002, 88, 41–48. [Google Scholar] [CrossRef]
- Marlier, Q.; D’Aes, T.; Verteneuil, S.; Vandenbosch, R.; Malgrange, B. Core cell cycle machinery is crucially involved in both life and death of post-mitotic neurons. Cell Mol. Life Sci. 2020, 77, 4553–4571. [Google Scholar] [CrossRef]
- Kurisu, S.; Takenawa, T. The WASP and WAVE family proteins. Genome Biol. 2009, 10, 226. [Google Scholar] [CrossRef]
- Hitzemann, R.; Iancu, O.D.; Reed, C.; Baba, H.; Lockwood, D.R.; Phillips, T.J. Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption. Brain Sci. 2019, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Covill-Cooke, C.; Howden, J.H.; Birsa, N.; Kittler, J.T. Ubiquitination at the mitochondria in neuronal health and disease. Neurochem. Int. 2018, 117, 55–64. [Google Scholar] [CrossRef]
- Sandebring, A.; Cedazo-Mínguez, A. Parkin—An E3 Ubiquitin Ligase with Multiple Substrates. Alz Dis. Parkinsonism 2012, S10:002. [Google Scholar] [CrossRef]
- Patel, J.; Panicker, N.; Dawson, V.L.; Dawson, T.M. Cell Biology of Parkin: Clues to the Development of New Therapeutics for Parkinson’s Disease. CNS Drugs 2022, 36, 1249–1267. [Google Scholar] [CrossRef] [PubMed]
- Cremer, J.N.; Amunts, K.; Schleicher, A.; Palomero-Gallagher, N.; Piel, M.; Rosch, F.; Zilles, K. Changes in the expression of neurotransmitter receptors in Parkin and DJ-1 knockout mice--A quantitative multireceptor study. Neuroscience 2015, 311, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88 Pt B, 179–188. [Google Scholar] [CrossRef]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef]
- Jha, R.K.; Kouzine, F.; Levens, D. MYC function and regulation in physiological perspective. Front. Cell Dev. Biol. 2023, 11, 1268275. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1alpha to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar]
- Hatazawa, Y.; Senoo, N.; Tadaishi, M.; Ogawa, Y.; Ezaki, O.; Kamei, Y.; Miura, S. Metabolomic Analysis of the Skeletal Muscle of Mice Overexpressing PGC-1alpha. PLoS ONE 2015, 10, e0129084. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metabol. 2015, 21, 95–108. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bazylianska, V.; Moszczynska, A. Parkin-deficient rats are resistant to neurotoxicity of chronic high-dose methamphetamine. Exp. Neurol. 2021, 345, 113811. [Google Scholar] [CrossRef]
- Bazylianska, V.; Sharma, A.; Chauhan, H.; Schneider, B.; Moszczynska, A. Dopamine and Methamphetamine Differentially Affect Electron Transport Chain Complexes and Parkin in Rat Striatum: New Insight into Methamphetamine Neurotoxicity. Int. J. Mol. Sci. 2021, 23, 363. [Google Scholar] [CrossRef]
- Hebron, M.; Chen, W.; Miessau, M.J.; Lonskaya, I.; Moussa, C.E. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem. 2014, 129, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Okarmus, J.; Havelund, J.F.; Ryding, M.; Schmidt, S.I.; Bogetofte, H.; Heon-Roberts, R.; Wade-Martins, R.; Cowley, S.A.; Ryan, B.J.; Faergeman, N.J.; et al. Identification of bioactive metabolites in human iPSC-derived dopaminergic neurons with PARK2 mutation: Altered mitochondrial and energy metabolism. Stem Cell Rep. 2021, 16, 1510–1526. [Google Scholar] [CrossRef]
- Pacelli, C.; Latorre, D.; Cocco, T.; Capuano, F.; Kukat, C.; Seibel, P.; Villani, G. Tight control of mitochondrial membrane potential by cytochrome c oxidase. Mitochondrion 2011, 11, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Dalmonte, M.E.; Forte, E.; Genova, M.L.; Giuffre, A.; Sarti, P.; Lenaz, G. Control of respiration by cytochrome c oxidase in intact cells: Role of the membrane potential. J. Biol. Chem. 2009, 284, 32331–32335. [Google Scholar] [CrossRef]
- Villani, G.; Greco, M.; Papa, S.; Attardi, G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998, 273, 31829–31836. [Google Scholar] [CrossRef]
- Pham, L.; Arroum, T.; Wan, J.; Pavelich, L.; Bell, J.; Morse, P.T.; Lee, I.; Grossman, L.I.; Sanderson, T.H.; Malek, M.H.; et al. Regulation of mitochondrial oxidative phosphorylation through tight control of cytochrome c oxidase in health and disease—Implications for ischemia/reperfusion injury, inflammatory diseases, diabetes, and cancer. Redox Biol. 2024, 78, 103426. [Google Scholar] [CrossRef]
- Kumar, M.; Acevedo-Cintron, J.; Jhaldiyal, A.; Wang, H.; Andrabi, S.A.; Eacker, S.; Karuppagounder, S.S.; Brahmachari, S.; Chen, R.; Kim, H.; et al. Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem Cell Rep. 2020, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Damiano, M.; Gautier, C.A.; Bulteau, A.L.; Ferrando-Miguel, R.; Gouarne, C.; Paoli, M.G.; Pruss, R.; Auchere, F.; L’Hermitte-Stead, C.; Bouillaud, F.; et al. Tissue- and cell-specific mitochondrial defect in Parkin-deficient mice. PLoS ONE 2014, 9, e99898. [Google Scholar] [CrossRef]
- Giguere, N.; Pacelli, C.; Saumure, C.; Bourque, M.J.; Matheoud, D.; Levesque, D.; Slack, R.S.; Park, D.S.; Trudeau, L.E. Comparative analysis of Parkinson’s disease-associated genes in mice reveals altered survival and bioenergetics of Parkin-deficient dopamine neurons. J. Biol. Chem. 2018, 293, 9580–9593. [Google Scholar] [CrossRef]
- Zanon, A.; Rakovic, A.; Blankenburg, H.; Doncheva, N.T.; Schwienbacher, C.; Serafin, A.; Alexa, A.; Weichenberger, C.X.; Albrecht, M.; Klein, C.; et al. Profiling of Parkin-binding partners using tandem affinity purification. PLoS ONE 2013, 8, e78648. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Yang, W.; Yang, R.; Luo, J.; He, L.; Liu, J.; Zhang, J. Increased Absolute Glutamate Concentrations and Glutamate-to-Creatine Ratios in Patients with Methamphetamine Use Disorders. Front. Psych. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, J.J.; Nam, H.J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.Y.; Ham, H.; Evans, D.; Kim, S.H.; et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol. Cell 2015, 60, 21–34. [Google Scholar] [CrossRef]
- Lane, J.; Martin, T.; Weeks, H.P.; Jiang, W.G. Structure and role of WASP and WAVE in Rho GTPase signalling in cancer. Cancer Gen. Prot. 2014, 11, 155–165. [Google Scholar]
- Campellone, K.G.; Lebek, N.M.; King, V.L. Branching out in different directions: Emerging cellular functions for the Arp2/3 complex and WASP-family actin nucleation factors. Eur. J. Cell Biol. 2023, 102, 151301. [Google Scholar]
- Scheyer, A.F.; Loweth, J.A.; Christian, D.T.; Uejima, J.; Rabei, R.; Le, T.; Dolubizno, H.; Stefanik, M.T.; Murray, C.H.; Sakas, C.; et al. AMPA Receptor Plasticity in Accumbens Core Contributes to Incubation of Methamphetamine Craving. Biol. Psych. 2016, 80, 661–670. [Google Scholar] [CrossRef]
- Young, E.J.; Aceti, M.; Griggs, E.M.; Fuchs, R.A.; Zigmond, Z.; Rumbaugh, G.; Miller, C.A. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol. Psych. 2014, 75, 96–104. [Google Scholar] [CrossRef]
- Pandey, S.; Miller, C.A. Targeting the cytoskeleton as a therapeutic approach to substance use disorders. Pharmacol. Res. 2024, 202, 107143. [Google Scholar] [CrossRef]
- Akagawa, R.; Nabeshima, Y.I.; Kawauchi, T. Alternative Functions of Cell Cycle-Related and DNA Repair Proteins in Post-mitotic Neurons. Front. Cell Dev. Biol. 2021, 9, 753175. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem. Biol. Interact. 2015, 234, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Simone, P.D.; Pavlov, Y.I.; Borgstahl, G.E.O. ITPA (inosine triphosphate pyrophosphatase): From surveillance of nucleotide pools to human disease and pharmacogenetics. Mut. Res. 2013, 753, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Rajgor, D.; Hanley, J.G. Differential regulation of the Rac1 GTPase-activating protein (GAP) BCR during oxygen/glucose deprivation in hippocampal and cortical neurons. J. Biol. Chem. 2017, 292, 20173–20183. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126 Pt 17, 3799–3804. [Google Scholar] [CrossRef]
- Dietrich, C.J.; Morad, M. Synaptic acidification enhances GABAA signaling. J. Neurosci. 2010, 30, 16044–16052. [Google Scholar] [CrossRef]
- Makani, S.; Chesler, M. Endogenous alkaline transients boost postsynaptic NMDA receptor responses in hippocampal CA1 pyramidal neurons. J. Neurosci. 2007, 27, 7438–7446. [Google Scholar] [CrossRef]
- Ouzzine, M.; Gulberti, S.; Ramalanjaona, N.; Magdalou, J.; Fournel-Gigleux, S. The UDP-glucuronosyltransferases of the blood-brain barrier: Their role in drug metabolism and detoxication. Front. Cell Neurosci. 2014, 8, 349. [Google Scholar] [CrossRef]
- Singh, N.; Nandy, S.K.; Jyoti, A.; Saxena, J.; Sharma, A.; Siddiqui, A.J.; Sharma, L. Protein Kinase C (PKC) in Neurological Health: Implications for Alzheimer’s Disease and Chronic Alcohol Consumption. Brain Sci. 2024, 14, 554. [Google Scholar] [CrossRef]
- Colgan, L.A.; Hu, M.; Misler, J.A.; Parra-Bueno, P.; Moran, C.M.; Leitges, M.; Yasuda, R. PKCalpha integrates spatiotemporally distinct Ca(2+) and autocrine BDNF signaling to facilitate synaptic plasticity. Nat. Neurosci. 2018, 21, 1027–1037. [Google Scholar] [CrossRef]
- Bell, J.D.; Park, E.; Ai, J.; Baker, A.J. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009, 16, 1665–1680. [Google Scholar] [CrossRef]
- Joch, M.; Ase, A.R.; Chen, C.X.; MacDonald, P.A.; Kontogiannea, M.; Corera, A.T.; Brice, A.; Seguela, P.; Fon, E.A. Parkin-mediated monoubiquitination of the PDZ protein PICK1 regulates the activity of acid-sensing ion channels. Mol. Biol. Cell 2007, 18, 3105–3118. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Wang, Y.T. GluA1-homomeric AMPA receptor in synaptic plasticity and neurological diseases. Neuropharmacology 2021, 197, 108708. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yuan, W.; Li, M.D. Genes and pathways co-associated with the exposure to multiple drugs of abuse, including alcohol, amphetamine/methamphetamine, cocaine, marijuana, morphine, and/or nicotine: A review of proteomics analyses. Mol. Neurobiol. 2011, 44, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Sadat-Shirazi, M.S.; Soltani, H.; Nikpour, N.; Haghshenas, M.; Khalifeh, S.; Mokri, A.; Zarrindast, M.R. Alteration of orexin-A and PKCalpha in the postmortem brain of pure-opioid and multi-drug abusers. Neuropeptides 2020, 83, 102074. [Google Scholar] [CrossRef]
- Thanos, P.K.; Kim, R.; Delis, F.; Ananth, M.; Chachati, G.; Rocco, M.J.; Masad, I.; Muniz, J.A.; Grant, S.C.; Gold, M.S.; et al. Chronic Methamphetamine Effects on Brain Structure and Function in Rats. PLoS ONE 2016, 11, e0155457. [Google Scholar] [CrossRef]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; Wong, C.; et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am. J. Psych. 2001, 158, 383–389. [Google Scholar] [CrossRef]
- Berman, S.M.; Voytek, B.; Mandelkern, M.A.; Hassid, B.D.; Isaacson, A.; Monterosso, J.; Miotto, K.; Ling, W.; London, E.D. Changes in cerebral glucose metabolism during early abstinence from chronic methamphetamine abuse. Mol. Psychiatry 2008, 13, 897–908. [Google Scholar] [CrossRef]
- London, E.D.; Simon, S.L.; Berman, S.M.; Mandelkern, M.A.; Lichtman, A.M.; Bramen, J.; Shinn, A.K.; Miotto, K.; Learn, J.; Dong, Y.; et al. Mood disturbances and regional cerebral metabolic abnormalities in recently abstinent methamphetamine abusers. Arch. Gen. Psych. 2004, 61, 73–84. [Google Scholar] [CrossRef]
- Ballesteros-Yanez, I.; Castillo, C.A.; Merighi, S.; Gessi, S. The Role of Adenosine Receptors in Psychostimulant Addiction. Front. Pharmacol. 2017, 8, 985. [Google Scholar] [CrossRef]
- Martinez, L.A.; Lees, M.E.; Ruskin, D.N.; Masino, S.A. A ketogenic diet diminishes behavioral responses to cocaine in young adult male and female rats. Neuropharmacology 2019, 149, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Feier, G.; Valvassori, S.S.; Varela, R.B.; Resende, W.R.; Bavaresco, D.V.; Morais, M.O.; Scaini, G.; Andersen, M.L.; Streck, E.L.; Quevedo, J. Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol. Biochem. Behav. 2013, 103, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Song, S.H.; Kim, S.; Jang, W.J.; Ryu, I.S.; Jeong, C.H.; Lee, S. Exploring the progression of drug dependence in a methamphetamine self-administration rat model through targeted and non-targeted metabolomics analyses. Sci. Rep. 2024, 14, 22543. [Google Scholar] [CrossRef] [PubMed]
- Berro, L.F.; Andersen, M.L.; Tufik, S.; Howell, L.L. GABAA receptor positive allosteric modulators modify the abuse-related behavioral and neurochemical effects of methamphetamine in rhesus monkeys. Neuropharmacology 2017, 123, 299–309. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Ke, Y.; Huang, X.; Chen, S.; Zhuang, D.; Zhou, Y.; Wu, M.; Wang, Y.; Lai, M.; et al. Ibudilast-Mediated Suppression of Neuronal TLR4 in the Prefrontal Cortex Mitigates Methamphetamine-Induced Neuroinflammation and Addictive Behaviours. Addict. Biol. 2025, 30, e70033. [Google Scholar] [CrossRef]
- Yang, H.; Hu, B.; Wang, X.; Chen, W.; Zhou, H. The effects of hyaluronan and proteoglycan link protein 1 (HAPLN1) in ameliorating spinal cord injury mediated by Nrf2. Biotechnol. Appl. Biochem. 2024, 71, 929–939. [Google Scholar] [CrossRef]
- Van den Oever, M.C.; Lubbers, B.R.; Goriounova, N.A.; Li, K.W.; Van der Schors, R.C.; Loos, M.; Riga, D.; Wiskerke, J.; Binnekade, R.; Stegeman, M.; et al. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology 2010, 35, 2120–2133. [Google Scholar] [CrossRef]
- Weber, K.T.; Swamynathan, S.K.; Guntaka, R.V.; Sun, Y. Angiotensin II and extracellular matrix homeostasis. Int. J. Biochem. Cell Biol. 1999, 31, 395–403. [Google Scholar] [CrossRef]
- Kobiec, T.; Otero-Losada, M.; Chevalier, G.; Udovin, L.; Bordet, S.; Menendez-Maissonave, C.; Capani, F.; Perez-Lloret, S. The Renin-Angiotensin System Modulates Dopaminergic Neurotransmission: A New Player on the Scene. Front. Synaptic Neurosci. 2021, 13, 638519. [Google Scholar] [CrossRef]
- Young, C.N.; Davisson, R.L. Angiotensin-II, the Brain, and Hypertension: An Update. Hypertension 2015, 66, 920–926. [Google Scholar] [CrossRef]
- Marchese, N.A.; Occhieppo, V.B.; Basmadjian, O.M.; Casarsa, B.S.; Baiardi, G.; Bregonzio, C. Angiotensin II modulates amphetamine-induced glial and brain vascular responses, and attention deficit via angiotensin type 1 receptor: Evidence from brain regional sensitivity to amphetamine. Eur. J. Neurosci. 2020, 51, 1026–1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, J.; Li, X.; Cui, Y.; Mao, Z.; Wu, B.; Xu, H.; Zhou, W.; Liu, Y. Inhibition of Methamphetamine Self-Administration and Reinstatement by Central Blockade of Angiotensin II Receptor in Rats. J. Pharmacol. Exp. Ther. 2019, 369, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Wilson, W.L.; Wakeling, V.; Boydstun, A.S.; Jensen, A.; Kawas, L.; Harding, J.W. The Hepatocyte Growth Factor/c-Met Antagonist, Divalinal-Angiotensin IV, Blocks the Acquisition of Methamphetamine Dependent Conditioned Place Preference in Rats. Brain Sci. 2012, 2, 298–318. [Google Scholar] [CrossRef]
- Ham, S.J.; Yoo, H.; Woo, D.; Lee, D.H.; Park, K.S.; Chung, J. PINK1 and Parkin regulate IP(3)R-mediated ER calcium release. Nat. Commun. 2023, 14, 5202. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Mizuno, K.; Ohkuma, S. Possible involvement of type 1 inositol 1,4,5-trisphosphate receptors up-regulated by dopamine D1 and D2 receptors in mouse nucleus accumbens neurons in the development of methamphetamine-induced place preference. Neuroscience 2012, 227, 22–29. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, H.; Sun, Y.; Gao, Z.; Sun, C.; Wei, F.; Gao, D. Gabrb2 knock-out mice exhibit double-directed PMDD-like symptoms: GABAAR subunits, neurotransmitter metabolism disruption, and allopregnanolone binding. Aging 2022, 14, 8437–8447. [Google Scholar] [CrossRef]
- Yates, J.R. Pharmacological Treatments for Methamphetamine Use Disorder: Current Status and Future Targets. Subst. Abuse Rehabil. 2024, 15, 125–161. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Size | Matches | ES | abs(ES) | NES | p | q |
|---|---|---|---|---|---|---|---|
| REACTOME_THE_CITRIC_ACID_TCA_CYCLE_ AND_RESPIRATORY_ELECTRON_TRANSPORT | 178 | 74 | −0.63798 | 0.637986 | −2.3925 | 0 | 0 |
| REACTOME_RESPIRATORY_ELECTRON_ TRANSPORT_ATP_SYNTHESIS_BY_ CHEMIOSMOTIC_COUPLING_AND_HEAT_ PRODUCTION_BY_UNCOUPLING_PROTEINS | 127 | 44 | −0.67383 | 0.673836 | −2.2669 | 0 | 0.004754 |
| KEGG_PARKINSONS_DISEASE | 130 | 47 | −0.63380 | 0.633808 | −2.1624 | 0 | 0.020338 |
| KEGG_OXIDATIVE_PHOSPHORYLATION | 132 | 42 | −0.65043 | 0.65043 | −2.1719 | 0 | 0.022539 |
| KEGG_HUNTINGTONS_DISEASE | 182 | 64 | −0.59313 | 0.59313 | −2.1477 | 0 | 0.024089 |
| GOBP_SECONDARY_METABOLIC_PROCESS | 56 | 11 | −0.85764 | 0.85764 | −2.0945 | 0 | 0.027277 |
| WP_ELECTRON_TRANSPORT_CHAIN_ OXPHOS_SYSTEM_IN_MITOCHONDRIA | 106 | 37 | −0.65395 | 0.65395 | −2.1107 | 0 | 0.028262 |
| REACTOME_RESPIRATORY_ ELECTRON_TRANSPORT | 103 | 31 | −0.67907 | 0.67907 | −2.095 | 0 | 0.029688 |
| WP_METAPATHWAY_ BIOTRANSFORMATION_PHASE_I_AND_II | 185 | 24 | −0.70232 | 0.70232 | −2.1020 | 0 | 0.029817 |
| GOBP_TRICARBOXYLIC_ACID_CYCLE | 34 | 24 | −0.70233 | 0.70233 | −2.1139 | 0 | 0.031092 |
| GOBP_CELLULAR_RESPIRATION | 187 | 62 | −0.58517 | 0.58517 | −2.1213 | 0.001832 | 0.03328 |
| GOBP_OXIDATIVE_PHOSPHORYLATION | 148 | 46 | −0.59655 | 0.59655 | −2.0551 | 0 | 0.046927 |
| REACTOME_PYRUVATE_METABOLISM_ AND_CITRIC_ACID_TCA_CYCLE | 55 | 32 | −0.63985 | 0.63985 | −2.0437 | 0 | 0.047845 |
| GOBP_ELECTRON_TRANSPORT_CHAIN | 178 | 50 | −0.59981 | 0.59981 | −2.0451 | 0 | 0.049982 |
| Name | Size | Matches | ES | abs(ES) | NES | p | q |
|---|---|---|---|---|---|---|---|
| GO_RNA_SPLICING_VIA_ TRANSESTERIFICATION_REACTIONS | 267 | 45 | 0.71708 | 0.71708 | 2.34074 | 0 | 0 |
| GO_RNA_SPLICING | 367 | 52 | 0.695816 | 0.695816 | 2.33113 | 0 | 0 |
| GO_RNA_PROCESSING | 835 | 138 | 0.595748 | 0.595748 | 2.29978 | 0 | 0 |
| GO_MRNA_PROCESSING | 432 | 58 | 0.668237 | 0.668237 | 2.27658 | 0 | 0 |
| GO_MRNA_METABOLIC_PROCESS | 611 | 136 | 0.553283 | 0.553283 | 2.1398 | 0 | 0.000381 |
| mRNA Splicing-Major Pathway | 106 | 22 | 0.759172 | 0.759172 | 2.13459 | 0 | 0.000272 |
| mRNA Splicing | 106 | 22 | 0.759172 | 0.759172 | 2.13459 | 0 | 0.000272 |
| mRNA Processing | 156 | 26 | 0.730662 | 0.730662 | 2.13292 | 0 | 0.000211 |
| Processing of Capped Intron-Containing Pre-mRNA | 137 | 26 | 0.730662 | 0.730662 | 2.13292 | 0 | 0.000211 |
| Gene Expression | 1082 | 190 | 0.514697 | 0.514697 | 2.06178 | 0 | 0.002379 |
| KEGG_SPLICEOSOME | 128 | 23 | 0.722244 | 0.722244 | 2.05753 | 0 | 0.002162 |
| GO_OSSIFICATION | 251 | 33 | 0.642403 | 0.642403 | 1.99171 | 0 | 0.005629 |
| GO_RIBONUCLEOPROTEIN_ COMPLEX_BIOGENESIS | 440 | 104 | 0.529386 | 0.529386 | 1.97458 | 0 | 0.007684 |
| Metabolism of mRNA | 220 | 118 | 0.514172 | 0.514172 | 1.94801 | 0 | 0.010534 |
| GO_NCRNA_PROCESSING | 386 | 81 | 0.534853 | 0.534853 | 1.93834 | 0 | 0.012812 |
| Thromboxane signaling through TP receptor | 23 | 9 | 0.86424 | 0.86424 | 1.92381 | 0 | 0.015104 |
| GO_RIBOSOMAL_SMALL_ SUBUNIT_BIOGENESIS | 59 | 13 | 0.785658 | 0.785658 | 1.91994 | 0 | 0.015167 |
| GO_SPLICEOSOMAL_COMPLEX_ASSEMBLY | 53 | 9 | 0.831624 | 0.831624 | 1.91816 | 0.003135 | 0.014694 |
| Metabolism of RNA | 266 | 124 | 0.503464 | 0.503464 | 1.91488 | 0 | 0.014772 |
| GO_OXIDOREDUCTION_COENZYME_ METABOLIC_PROCESS | 107 | 45 | 0.58676 | 0.58676 | 1.90271 | 0 | 0.018743 |
| GO_NUCLEAR_TRANSCRIBED_MRNA_ CATABOLIC_PROCESS_NONSENSE_ MEDIATED_DECAY | 118 | 75 | 0.522073 | 0.522073 | 1.86156 | 0 | 0.03783 |
| GO_OSTEOBLAST_DIFFERENTIATION | 126 | 25 | 0.635636 | 0.635636 | 1.85942 | 0 | 0.037321 |
| L13a-mediated translational silencing of Ceruloplasmin expression | 104 | 82 | 0.512746 | 0.512746 | 1.85683 | 0 | 0.03548 |
| 3′-UTR-mediated translational regulation | 104 | 82 | 0.512746 | 0.512746 | 1.85683 | 0 | 0.03548 |
| Formation of a pool of free 40S subunits | 94 | 76 | 0.517639 | 0.517639 | 1.85581 | 0 | 0.034441 |
| GO_RRNA_METABOLIC_PROCESS | 255 | 72 | 0.517731 | 0.517731 | 1.8484 | 0 | 0.037178 |
| GO_RIBOSOME_BIOGENESIS | 308 | 77 | 0.513105 | 0.513105 | 1.84816 | 0 | 0.035872 |
| Nonsense-Mediated Decay | 106 | 74 | 0.518657 | 0.518657 | 1.84526 | 0 | 0.035071 |
| Nonsense Mediated Decay Enhanced by the Exon Junction Complex | 106 | 74 | 0.518657 | 0.518657 | 1.84526 | 0 | 0.035071 |
| GO_REGULATION_OF_RNA_SPLICING | 97 | 24 | 0.641889 | 0.641889 | 1.83829 | 0.001427 | 0.037359 |
| GO_TRANSLATIONAL_INITIATION | 146 | 87 | 0.502547 | 0.502547 | 1.83806 | 0 | 0.03643 |
| Signal amplification | 32 | 13 | 0.746826 | 0.746826 | 1.83442 | 0 | 0.03761 |
| GO_NCRNA_METABOLIC_PROCESS | 533 | 88 | 0.501376 | 0.501376 | 1.83227 | 0 | 0.03872 |
| Presynaptic function of Kainate receptors | 21 | 7 | 0.870796 | 0.870796 | 1.83088 | 0 | 0.037241 |
| G beta:gamma signalling through PLC beta | 20 | 7 | 0.870796 | 0.870796 | 1.83088 | 0 | 0.037241 |
| Eukaryotic Translation Initiation | 112 | 85 | 0.503589 | 0.503589 | 1.82892 | 0 | 0.036488 |
| Cap-dependent Translation Initiation | 112 | 85 | 0.503589 | 0.503589 | 1.82892 | 0 | 0.036488 |
| GTP hydrolysis and joining of the 60S ribosomal subunit | 105 | 82 | 0.504021 | 0.504021 | 1.82533 | 0 | 0.038107 |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | 32 | 23 | 0.65316 | 0.65316 | 1.82266 | 0.001364 | 0.039618 |
| Eukaryotic Translation Elongation | 89 | 72 | 0.509709 | 0.509709 | 1.81343 | 0 | 0.044788 |
| Nonsense Mediated Decay Independent of the Exon Junction Complex | 89 | 69 | 0.51292 | 0.51292 | 1.81174 | 0 | 0.044832 |
| GO_RNA_CATABOLIC_PROCESS | 227 | 84 | 0.500861 | 0.500861 | 1.81094 | 0 | 0.044241 |
| Formation of the ternary complex, and subsequently, the 43S complex | 48 | 41 | 0.564116 | 0.564116 | 1.80663 | 0 | 0.045756 |
| GO_REGULATION_OF_CIRCADIAN_RHYTHM | 103 | 19 | 0.670194 | 0.670194 | 1.80448 | 0 | 0.04623 |
| Translation | 148 | 99 | 0.48732 | 0.48732 | 1.80395 | 0 | 0.045435 |
| GO_AEROBIC_RESPIRATION | 53 | 29 | 0.604936 | 0.604936 | 1.8022 | 0 | 0.045854 |
| Peptide chain elongation | 84 | 69 | 0.506827 | 0.506827 | 1.80094 | 0 | 0.045465 |
| Translation initiation complex formation | 55 | 45 | 0.550771 | 0.550771 | 1.79833 | 0 | 0.045279 |
| Activation of the mRNA upon binding of the cap-binding complex and eIFs, and subsequent binding to 43S | 56 | 45 | 0.550771 | 0.550771 | 1.79833 | 0 | 0.045279 |
| GO_TRICARBOXYLIC_ACID_METABOLIC_ PROCESS | 37 | 26 | 0.630346 | 0.630346 | 1.79827 | 0 | 0.044412 |
| ADP signalling through P2Y purinoceptor 1 | 25 | 9 | 0.801737 | 0.801737 | 1.79777 | 0 | 0.043989 |
| GO_CELLULAR_RESPIRATION | 143 | 58 | 0.535198 | 0.535198 | 1.79474 | 0 | 0.045668 |
| Eukaryotic Translation Termination | 84 | 68 | 0.509707 | 0.509707 | 1.79437 | 0 | 0.045004 |
| GO_RIBONUCLEOPROTEIN_COMPLEX_ LOCALIZATION | 118 | 18 | 0.661641 | 0.661641 | 1.79191 | 0.001458 | 0.045738 |
| KEGG_RIBOSOME | 88 | 66 | 0.50964 | 0.50964 | 1.78599 | 0 | 0.048781 |
| Name | Group | Size | Matches | ES | abs(ES) | NES | p | q |
|---|---|---|---|---|---|---|---|---|
| GOBP_AEROBIC_RESPIRATION | PKO | 86 | 39 | −0.571 | 0.571 | −1.899 | 0.00385 | 0.1238 |
| GOBP_AEROBIC_RESPIRATION | PO | 86 | 39 | 0.564 | 0.564 | 1.781 | 0 | 0.0516 |
| GOBP_CELLULAR_RESPIRATION | PKO | 187 | 62 | −0.585 | 0.585 | −2.121 | 0.00183 | 0.0333 |
| GOBP_CELLULAR_RESPIRATION | PO | 187 | 62 | 0.519 | 0.519 | 1.802 | 0 | 0.0467 |
| GOBP_ENERGY_DERIVATION_BY_ OXIDATION_OF_ORGANIC_ COMPOUNDS | PKO | 278 | 79 | −0.516 | 0.516 | −1.953 | 0 | 0.0835 |
| GOBP_ENERGY_DERIVATION_BY_ OXIDATION_OF_ORGANIC_ COMPOUNDS | PO | 278 | 79 | 0.454 | 0.454 | 1.628 | 0.00524 | 0.2103 |
| GOBP_POSITIVE_REGULATION_OF_ MITOTIC_CELL_CYCLE | PKO | 118 | 15 | −0.657 | 0.657 | −1.786 | 0.00986 | 0.2058 |
| GOBP_POSITIVE_REGULATION_OF_ MITOTIC_CELL_CYCLE | PO | 118 | 15 | 0.652 | 0.652 | 1.672 | 0.00884 | 0.1433 |
| GOBP_TRICARBOXYLIC_ACID_CYCLE | PKO | 34 | 24 | −0.702 | 0.702 | −2.114 | 0 | 0.0311 |
| GOBP_TRICARBOXYLIC_ACID_CYCLE | PO | 34 | 24 | 0.618 | 0.618 | 1.806 | 0.00145 | 0.0456 |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | PKO | 31 | 23 | −0.689 | 0.689 | −2.022 | 0.00197 | 0.0528 |
| KEGG_CITRATE_CYCLE_TCA_CYCLE | PO | 31 | 23 | 0.653 | 0.653 | 1.877 | 0 | 0.0263 |
| REACTOME_RHO_GTPASES_ACTIVATE_ WASPS_AND_WAVES | PKO | 36 | 20 | −0.638 | 0.638 | −1.813 | 0.01158 | 0.1819 |
| REACTOME_RHO_GTPASES_ACTIVATE_ WASPS_AND_WAVES | PO | 36 | 20 | 0.652 | 0.652 | 1.807 | 0.00287 | 0.0453 |
| WP_TCA_CYCLE_AKA_KREBS_OR_ CITRIC_ACID_CYCLE | PKO | 18 | 14 | −0.773 | 0.773 | −2.028 | 0.00190 | 0.0573 |
| WP_TCA_CYCLE_AKA_KREBS_OR_ CITRIC_ACID_CYCLE | PO | 18 | 14 | 0.632 | 0.632 | 1.621 | 0.02025 | 0.2179 |
| Symbol * | Entrez Gene Name | Gene Symbol—Rat | FC_PKO | p-Value PKO |
|---|---|---|---|---|
| AGT | angiotensinogen | Agt | 1.699 | 0.00153 |
| GSTO1 | glutathione S-transferase omega 1 | Gsto1 | −1.139 | 0.0226 |
| GSTP1 | glutathione S-transferase pi 1 | Gstp1 | −1.235 | 0.00321 |
| HAPLN1 | hyaluronan and proteoglycan link protein 1 | Hapln1 | −1.943 | 0.0428 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 | Nqo1 | −1.593 | 0.00605 |
| PI4KA | phosphatidylinositol 4-kinase α | Pi4ka | −1.044 | 0.00694 |
| Symbol * | Entrez Gene Name | Gene Symbol—Rat | FC_PO | p-value PO |
| GABRB2 | γ-aminobutyric acid type A receptor subunit beta 2 | Gabrb2 | −1.813 | 0.0191 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Atasi, T.; Collin, F.; Wang, W.; Lam, T.T.; Garcia-Milian, R.; Arroum, T.; Pham, L.; Hüttemann, M.; Moszczynska, A. The Proteomic Landscape of Parkin-Deficient and Parkin-Overexpressing Rat Nucleus Accumbens: An Insight into the Role of Parkin in Methamphetamine Use Disorder. Biomolecules 2025, 15, 958. https://doi.org/10.3390/biom15070958
Sharma A, Atasi T, Collin F, Wang W, Lam TT, Garcia-Milian R, Arroum T, Pham L, Hüttemann M, Moszczynska A. The Proteomic Landscape of Parkin-Deficient and Parkin-Overexpressing Rat Nucleus Accumbens: An Insight into the Role of Parkin in Methamphetamine Use Disorder. Biomolecules. 2025; 15(7):958. https://doi.org/10.3390/biom15070958
Chicago/Turabian StyleSharma, Akhil, Tarek Atasi, Florine Collin, Weiwei Wang, TuKiet T. Lam, Rolando Garcia-Milian, Tasnim Arroum, Lucynda Pham, Maik Hüttemann, and Anna Moszczynska. 2025. "The Proteomic Landscape of Parkin-Deficient and Parkin-Overexpressing Rat Nucleus Accumbens: An Insight into the Role of Parkin in Methamphetamine Use Disorder" Biomolecules 15, no. 7: 958. https://doi.org/10.3390/biom15070958
APA StyleSharma, A., Atasi, T., Collin, F., Wang, W., Lam, T. T., Garcia-Milian, R., Arroum, T., Pham, L., Hüttemann, M., & Moszczynska, A. (2025). The Proteomic Landscape of Parkin-Deficient and Parkin-Overexpressing Rat Nucleus Accumbens: An Insight into the Role of Parkin in Methamphetamine Use Disorder. Biomolecules, 15(7), 958. https://doi.org/10.3390/biom15070958