
Heterostilbene Carbamates with Selective and Remarkable Butyrylcholinesterase Inhibition: Computational Study and Physico-Chemical Properties

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. General Remarks
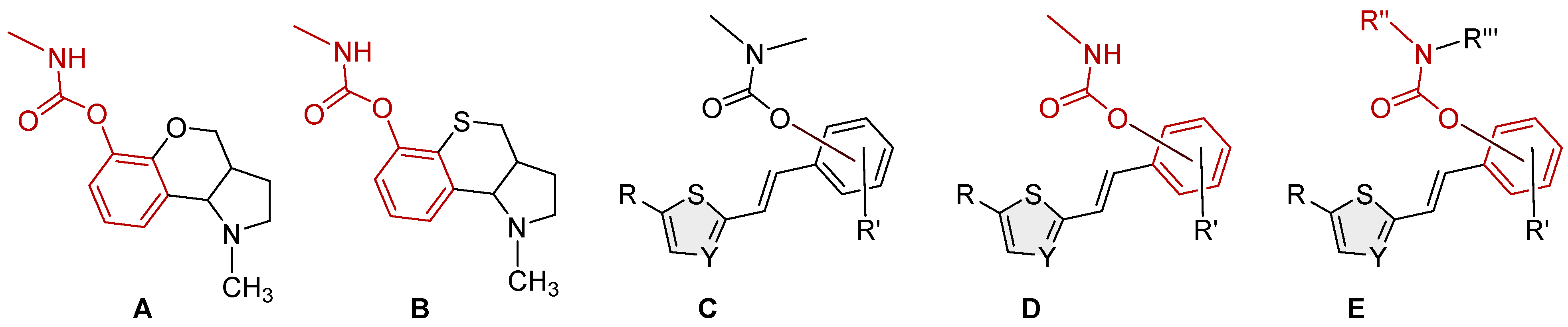
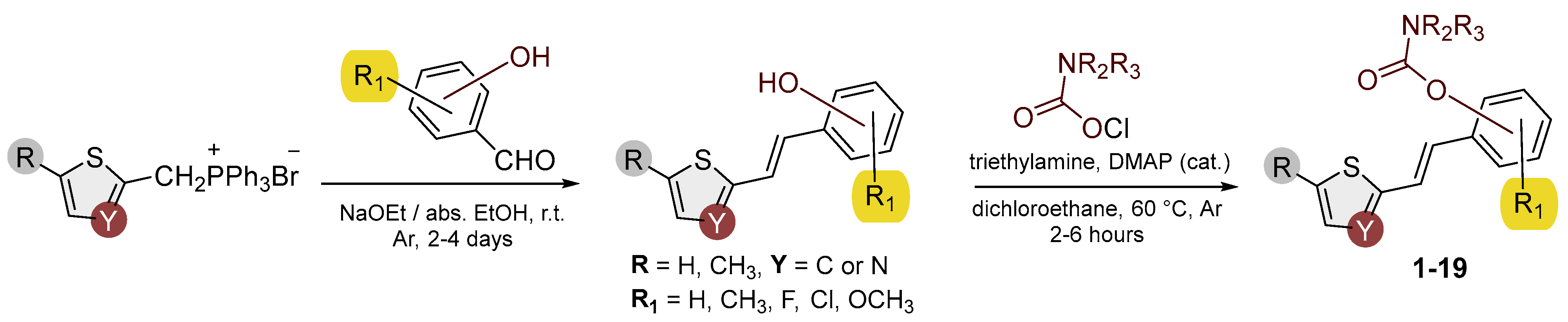
2.2. Synthesis of Heterostilbene Carbamates 1–19
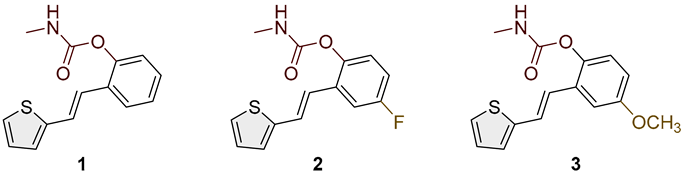
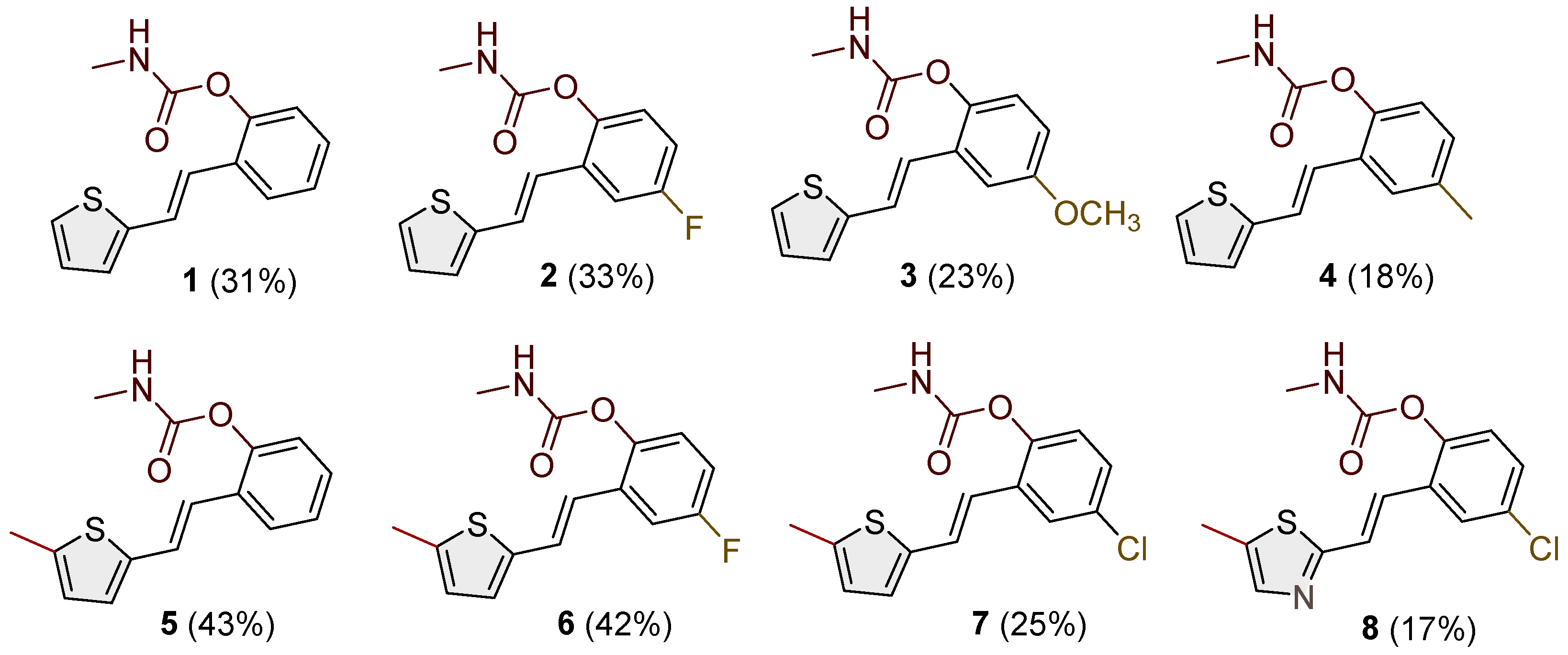
- (E)-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (1): 37 mg (isolated 31%), white powder; m.p. 117–119 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.61 (d, J = 7.4 Hz, 1H), 7.26 (m, 3H), 7.20 (d, J = 5.3 Hz, 2H), 7.13 (d, J = 17.5 Hz, 1H), 7.12–7.10 (m, 1H), 7.05–7.00 (m, 1H), 5.10 (br s, 1H), 2.92 (d, J = 4.8 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 155.0, 154.0, 148.3, 142.9, 128.3, 127.6, 126.4, 126.1, 125.8, 124.6, 123.6, 123.0, 121.8, 27.9; HRMS (ESI) (m/z) for C14H13NO2S: [M + H]+calcd = 259.0667, and [M + H]+measured = 259.0666.
- (E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (2): 39 mg (isolated 33%), white powder; m.p. 110–111 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.28 (d, J = 9.7, 1H), 7.22 (d, J = 4.5 Hz, 1H), 7.18 (d, J = 18.4 Hz, 1H), 7.09 –7.02 (m, 2H), 7.01 (t, J = 2.7 Hz, 1H), 7.00–6.9 (m, 2H), 5.10 (br s, 1H), 2.92 (d, J = 4.2, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 160.2 (d, JCF = 243.3 Hz), 155.1, 144.3, 142.0, 130.6 (d, JCF = 7.7 Hz), 127.7, 125.1, 124.6, 124.0 (d, JCF = 8.7 Hz), 120.7, 114.9, 111.9, 27.6; HRMS (ESI) (m/z) for C14H12FNO2S: [M + H]+calcd = 277.0573, and [M + H]+measured = 277.0572.
- (E)-4-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (3): 27 mg (isolated 23%), white powder; m.p. 123–124 °C; Rf (50% PE/E) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.33–7.21 (m, 2H), 7.20 (d, J = 3.1 Hz, 1H), 7.10 (d, J = 3.1 Hz, 1H), 7.03 (d, J = 8.9 Hz, 1H), 7.00 (t, J = 3.9 Hz, 1H), 6.91 (d, J = 16.3 Hz, 1H), 6.82 (dd, J = 2.7, 9.6 Hz, 1H), 5.00 (br s, 1H), 3.81 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 157.1, 155.4, 142.7, 142.2, 129.9, 130.7, 127.6, 126.5, 124.7, 123.8, 121.8, 114.1, 110.4, 55.7, 29.7; HRMS (ESI) (m/z) for C15H15NO3S: [M + H]+calcd = 289.0772, and [M + H]+measured = 289.0773.
![Biomolecules 15 00825 i002]()
- (E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (4): 22 mg (isolated 18%), white powder; m.p. 109–111 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.41 (s, 1H), 7.22–7.17 (m, 2H), 7.09–7.04 (m, 2H), 7.03–6.96 (m, 3H), 5.04 (br s, 1H), 2.92 (d, J = 4.8 Hz, 3H), 2.35 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 155.2, 146.2, 143.0, 135.3, 129.4, 129.1, 127.6, 126.4, 126.3, 124.5, 123.3, 122.7, 121.9, 27.9, 21.0; HRMS (ESI) (m/z) for C15H15NO2S: [M + H]+calcd = 273.0824, and [M + H]+measured = 273.0826.
- (E)-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (5): 18 mg (isolated 43%), white powder; m.p. 114–116 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.66–7.55 (m, 1H), 7.24–7.16 (m, 2H), 7.15–7. 09 (m, 2H), 6.89 (t, J = 16.7 Hz, 1H), 6.85 (d, J = 3.8 Hz, 1H), 6.60 (d, J = 3.5 Hz, 1H), 5.07 (br s, 1H), 2.93 (d, J = 4.6 Hz, 3H), 2.48 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 155.0, 148.2, 140.9, 139.5, 130.0, 128.0, 126.8, 125.9, 125.8, 125.7, 124.0, 123.0, 120.5, 29.7, 15.6; HRMS (ESI) (m/z) for C15H15NO2S: [M + H]+calcd = 273.0824, and [M + H]+measured = 273.0825.
- (E)-4-fluoro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (6): 19 mg (isolated 42%), white powder; m.p. 105–107 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.29–7.23 (m, 2H), 7.09 (d, J = 16.4 Hz, 2H), 6.87 (d, J = 3.4 Hz, 1H), 6.80 (d, J = 16.4 Hz, 1H), 6.66–6.64 (m, 1H), 5.06 (br s), 2.93 (d, J = 4.9 Hz, 3H), 2.48 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 160.3 (JCF = 243.1 Hz), 155.0, 144.0, 140.3, 140.2, 131.9 (d, JCF = 8.3 Hz), 127.5, 125.9, 125.0, 124.4 (d, JCF = 8.3 Hz), 119.4, 114.7 (d, JCF = 24.1 Hz), 111.8 (d, JCF = 24.1 Hz), 29.5, 15.7; HRMS (ESI) (m/z) for C15H14FNO2S: [M + H]+calcd = 291.0729, and [M + H]+measured = 291.0731.
![Biomolecules 15 00825 i003]()
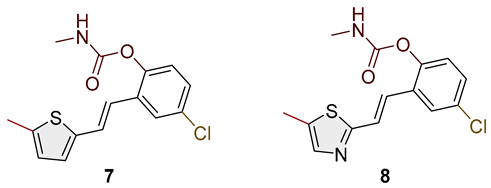
- (E)-4-chloro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (7): 12 mg (isolated 25%), white powder; m.p. 122–123 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.55 (d, J = 2.5 Hz, 1H), 7.13–7.09 (m, 2H), 7.06 (d, J = 8.9 Hz, 1H), 6.87 (d, J = 3.4, 1H), 6.80–6.76 (m, 1H), 6.66–6.64 (m, 1H), 5.07 (br s), 2.93 (d, J = 4.9 Hz, 3H), 2.47 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 154.7, 147.7, 146.5, 140.3, 131.8, 131.2, 127.7, 127.5, 125.9, 125.5, 125.2, 125.1, 124.3, 29.7, 15.7; HRMS (ESI) (m/z) for C15H14ClNO2S: [M + H]+calcd = 307.0434, and [M + H]+measured = 307.0433.
- Mixture of (E) and (Z)-4-chloro-2-(2-(5-methylthiazol-2-yl)vinyl)phenyl methylcarbamate (8); for (E)-isomer, the data are the following: 8 mg (isolated 17%), Rf (50% PE/E) = 0.30; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.52 (s, 1H), 7.26 (s, 1H), 7.12 (d, J = 16.6 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 16.3 Hz, 1H), 2.58 (d, J = 5.0 Hz, 3H), 2.57 (s, 3H); MS (ESI) (m/z) for C17H19NO3S: [M + H]+ 308 (100).
![Biomolecules 15 00825 i004]()
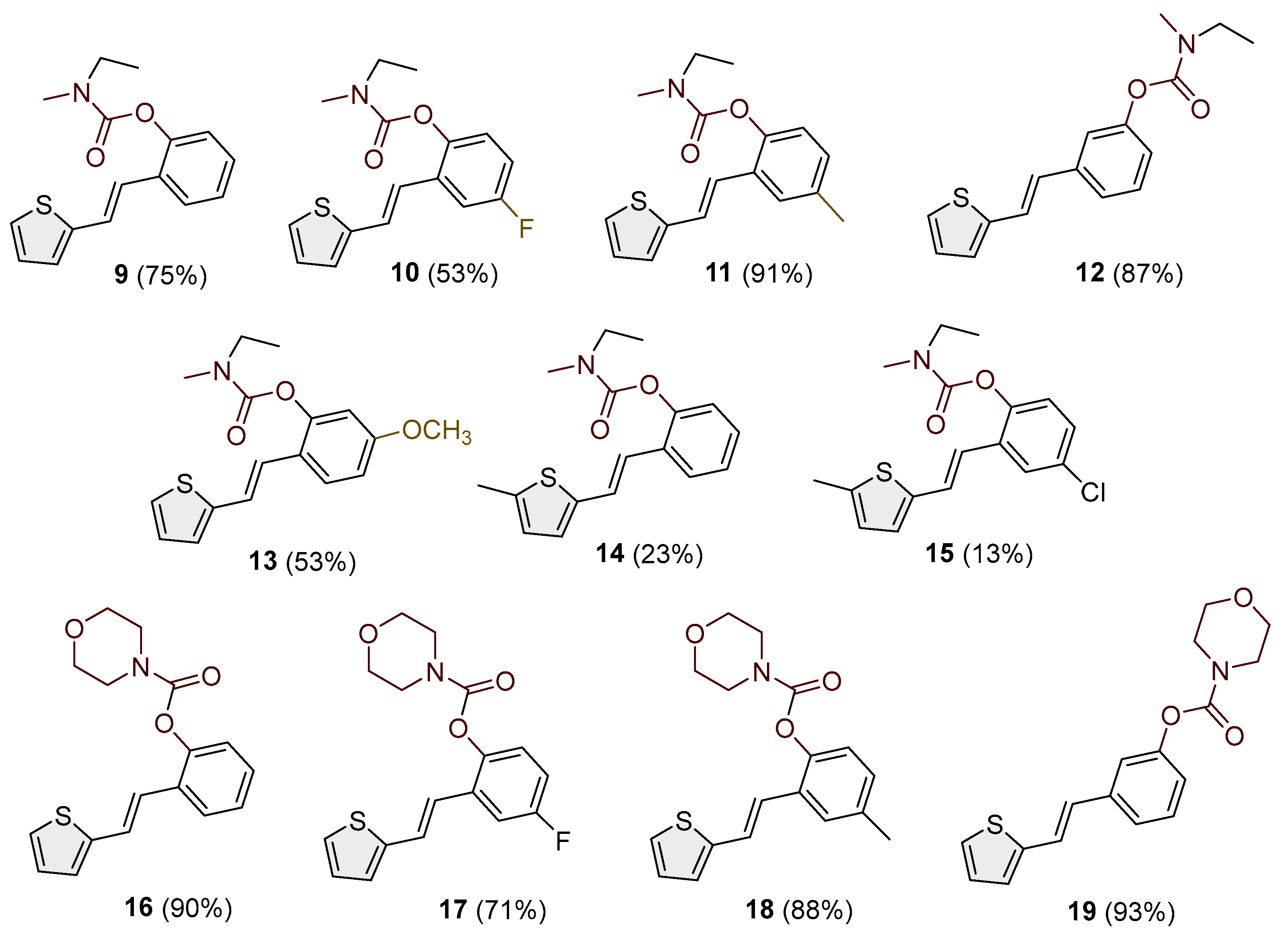
- (E)-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (9): 40 mg (isolated 75%), colorless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.63–7.58 (m, 1H), 7.38–7.31 (m, 1H), 7.27–7.23 (m, 1H), 7.22–7.17 (m, 3H), 7.13 (t, J = 7.15 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 7.00–6.98 (m, 1H), 3.50 (dq, J = 7.3, 80.6 Hz, 2H), 3.10 (d, J = 87.6 Hz, 3H), 1.28–1.22 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 154.3, 148.8, 143.1, 130.4, 129.7, 128.8, 128.3, 127.6, 126.3, 125.6, 124.5, 123.4, 122.8, 44.1, 21.0, 13.5; HRMS (ESI) (m/z) for C16H17NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0985.
- (E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (10): 28 mg (isolated 53%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.33–7.27 (m, 1H), 7.22 (d, J = 3.89 Hz, 1H), 7.17–7.04 (m, 3H), 7.03–6.86 (m, 3H), 3.49 (dq, J = 7.3, 39.1 Hz, 2H), 3.09 (d, J = 43.3 Hz, 3H), 1.25–1.20. (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1 (d, JCF = 242.6 Hz), 158.5, 154.1, 144.7, 142.5, 127.7, 127.1, 126.5, 126.3, 125.0 (d, JCF = 8.7 Hz), 120.8 (d, JCF = 8.7 Hz), 115.1 (d, JCF = 24.1 Hz), 111.8 (d, JCF = 24.1 Hz), 44.1, 34.5, 14.2.
- (E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (11): 85 mg (isolated 91%), colorless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.41 (s, 1H), 7.21–7.15 (m, 2H), 7.10–6.94 (m, 5H), 3.49 (dq, J = 7.0, 79.1 Hz, 2H), 3.08 (d, J = 86.4 Hz, 3H), 2.34 (s, 3H), 1.33–1.28 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: too small a quantity for recording; HRMS (ESI) (m/z) for C17H19NO2S: [M + H]+calcd = 301.1136, and [M + H]+measured = 301.1141.
![Biomolecules 15 00825 i005]()
- (E)-3-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (12): 63 mg (isolated 87%), colorless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.31 (t, J = 7.7 Hz, 1H), 7.28–7.22 (m, 3H), 7.20 (d, J = 4.5 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H), 7.03–6.97 (m, 2H), 6.90 (d, J = 15.8 Hz, 1H), 3.46 (dq, J = 7.2, 39.8 Hz, 2H), 3.04 (d, J = 45.6 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 154.5, 151.9, 142.6, 138.4, 129.4, 128.3, 127.6, 126.3, 124.5, 123.3, 122.5, 120.9, 119.2, 44.1, 34.3, 13.2; HRMS (ESI) (m/z) for C16H17NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0982.
- (E)-5-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (13): 18 mg (isolated 53%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.52 (d, J = 8.9 Hz, 1H), 7.16–7.03 (m, 2H), 7.02–6.84 (m, 3H), 6.80–6.66 (m, 2H), 3.81 (s, 3H), 3.50 (dq, J = 7.2, 37.7 Hz, 2H), 3.09 (d, J = 42.4 Hz, 3H), 1.26–1.20 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1, 159.8, 149.6, 143.4, 127.6, 126.4, 125.7, 123.8, 122.4, 121.7, 121.3, 112.4, 108.3, 55.5, 44.2, 29.7, 13.5; MS (ESI) (m/z) for C17H19NO3S: [M + H]+ 318 (100).
- (E)-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (14): 25 mg (isolated 23%), colorless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.58–7.50 (m, 1H), 7.16–7.11 (m, 1H), 7.05 (dt, J = 8.5; 2.1 Hz, 1H), 7.02–6.93 (m, 2H), 6.75 (d, J = 15.0 Hz, 1H), 6.39 (dd, J = 15.0; 1.8 Hz, 1H), 3.49 (dq, J = 7.9, 72.5 Hz, 2H), 3.05–2.84 (m, 3H), 2.60–2.32 (m, 3H), 1.24–1.18 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: too small a quantity for recording; MS (ESI) (m/z) for C17H19NO2S: [M + H]+ 302 (100).
![Biomolecules 15 00825 i006]()
- (E)-4-chloro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (15): 8 mg (isolated 13%), colorless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.20–7.14 (m, 2H), 7.07–6.96 (m, 5H), 3.50 (dq, J = 7.0, 79.1 Hz, 2H), 3.09 (d, J = 86.4 Hz, 3H), 2.35 (s, 3H), 1.31–1.27 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: too small a quantity for recording; HRMS (ESI) (m/z) for C17H18ClNO2S: [M + H]+calcd = 335.0747, and [M + H]+measured = 335.0744.
- (E)-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (16): 75 mg (isolated 90%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.62 (dd, J = 1.7, 7.5 Hz, 1H) 7.21–7.09 (m, 4H), 7.03–6.84 (m, 4H), 3.88–3. 44 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.4, 148.5, 142.9, 139.6, 130.4, 128.4, 127.7, 126.7, 125.9, 124.6, 123.1, 121.6, 66.8, 66.6 (one quaternary C is missing); HRMS (ESI) (m/z) for C17H17NO3S: [M + H]+calcd = 315.0929, and [M + H]+measured = 315.0932.
- (E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (17): 40 mg (isolated 71%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.33–7.20 (m, 3H), 7.16–6.92 (m, 4H), 6.88 (d, J = 15.5 Hz, 1H), 3.88–3.43 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.3 (d, J = 244.3 Hz), 153.3, 144.3, 142.3, 131.4 (d, J = 8.5 Hz), 127.8, 127.3, 126.5 (d, J = 8.5 Hz), 125.1, 124.8, 120.5, 115.0 (d, J = 23.9 Hz), 111.9 (d, J = 23.9 Hz), 66.7, 66.6; HRMS (ESI) (m/z) for C17H16FNO3S: [M + H]+calcd = 333.0835, and [M + H]+measured = 333.0838.
![Biomolecules 15 00825 i007]()
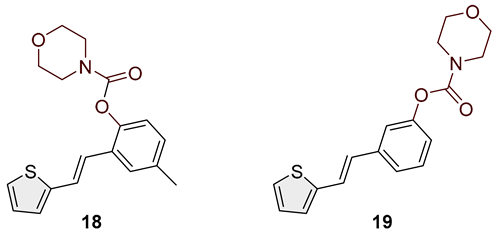
- (E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (18): 78 mg (isolated 88%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.41 (s, 1H), 7.26–7.14 (m, 2H), 7.11–7.00 (m, 4H), 6.93 (d, J = 16.3 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.6, 146.4, 143.0, 135.4, 129.2, 127.7, 126.5, 126.4, 124.5, 123.4, 122.7, 121.7, 66.7, 66.5, 21.0 (one quaternary C is missing); HRMS (ESI) (m/z) for C18H19NO3S: [M + H]+calcd = 329.1086, and [M + H]+measured = 329.1091.
- (E)-3-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (19): 87 mg (isolated 93%), colorless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.38–7.28 (m, 2H), 7.25–7.18 (m, 3H), 7.14–6.95 (m, 3H), 6.89 (d, J = 16.0 Hz, 1H), 3.80–3.50 (m, 8H) 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.6, 151.6, 142.6, 138.6, 129.5, 127.6, 127.4, 126.4, 124.6, 120.7, 119.1, 66.6, 66.5 (two quaternary C are missing); HRMS (ESI) (m/z) for C22H23NO3S: [M + H]+calcd = 315.0929, and [M + H]+measured = 315.0934.




2.3. X-Ray Crystallography
2.4. In Vitro ChE Activity Assay
2.5. Anti-Inflammatory Activity
2.6. Computational Details
2.7. ADME-Tox Predictions
3. Results and Discussion
3.1. Synthesis and Characterization of New Heterostilbene Carbamates 1–19
3.2. Crystal Structures of Carbamates 1 and 4
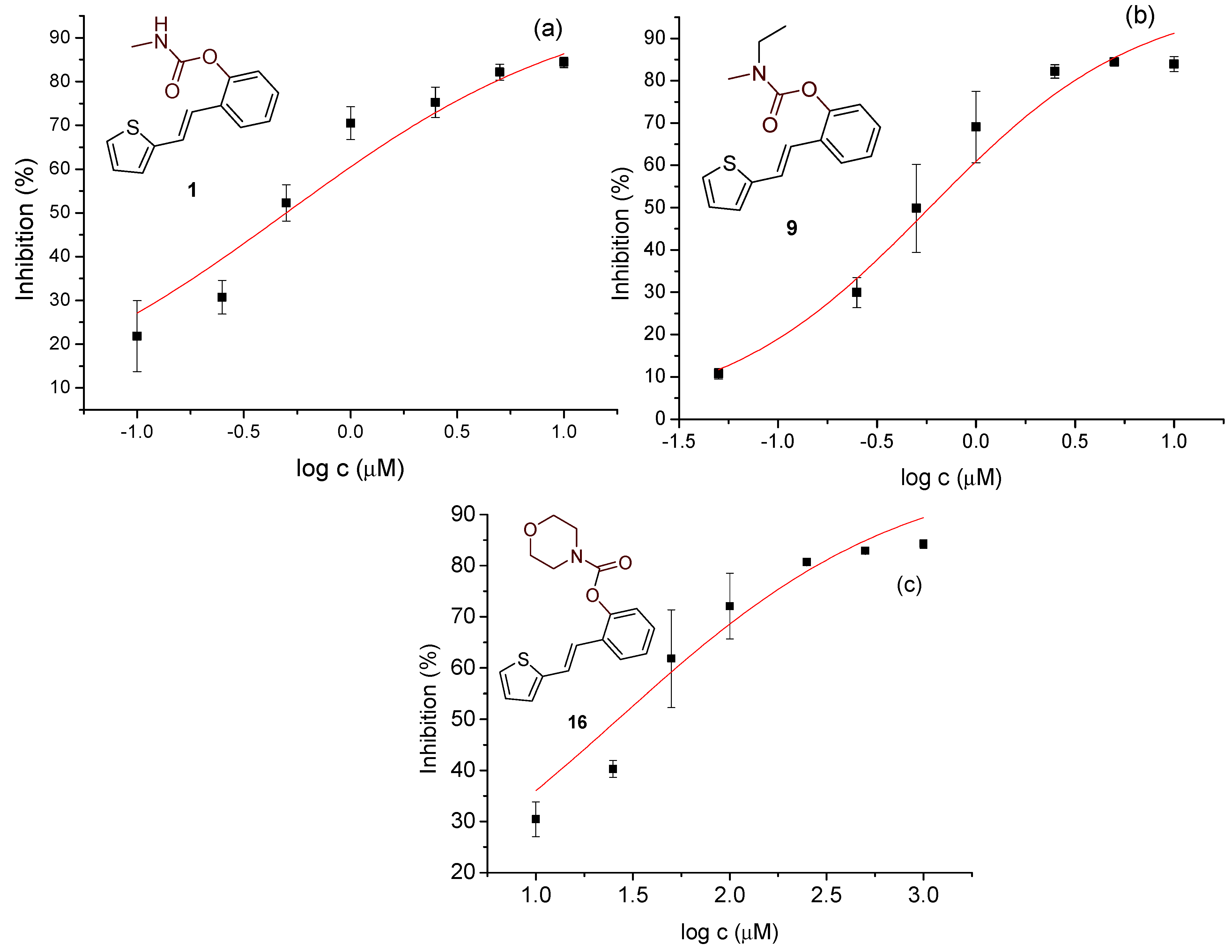
3.3. Cholinesterase Inhibition of Heterostilbene Carbamates 1–19
3.4. Anti-Inflammatory Activity of Heterostilbene Carbamates 1–19
3.5. ADME(T) Properties and Genotoxicity—ICH (M7) Q(SAR)
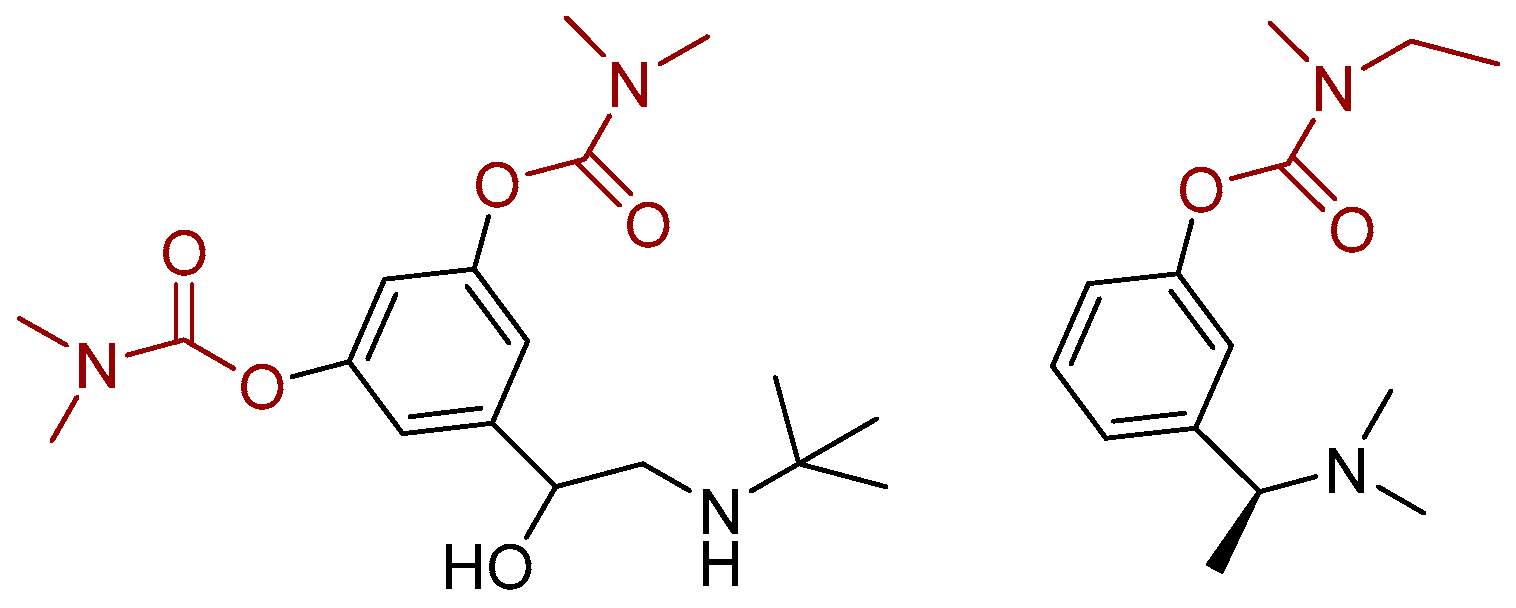
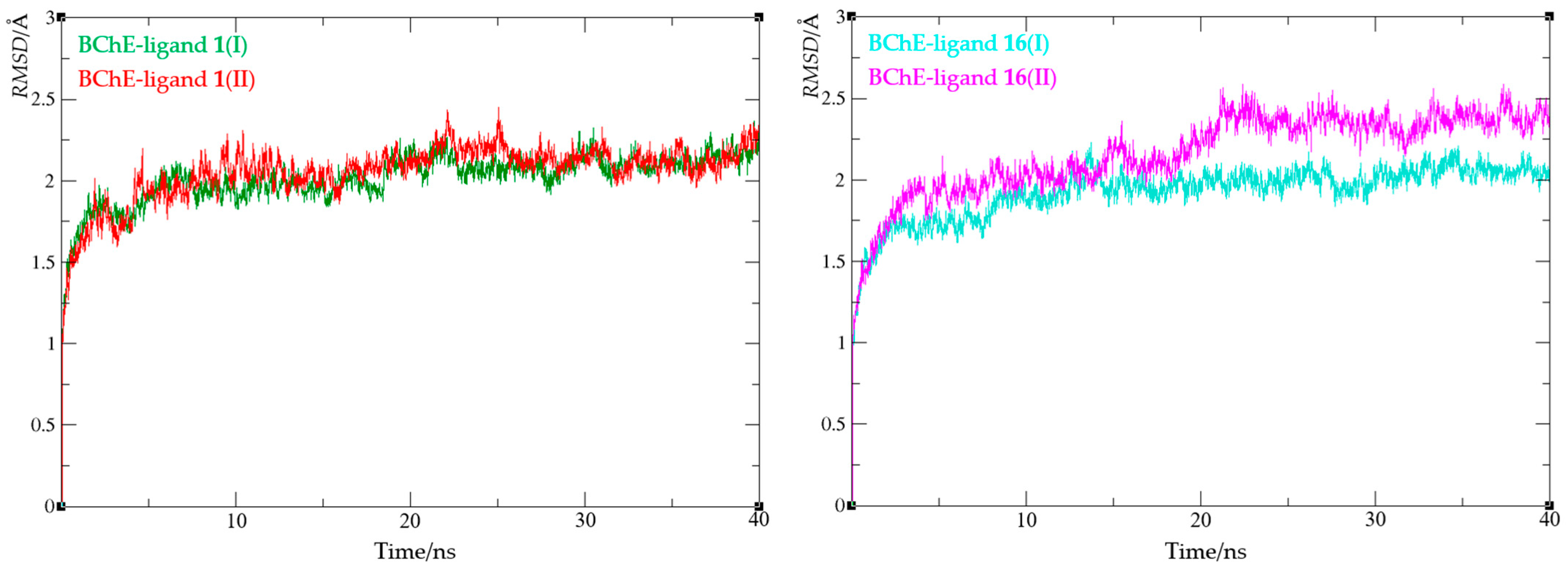
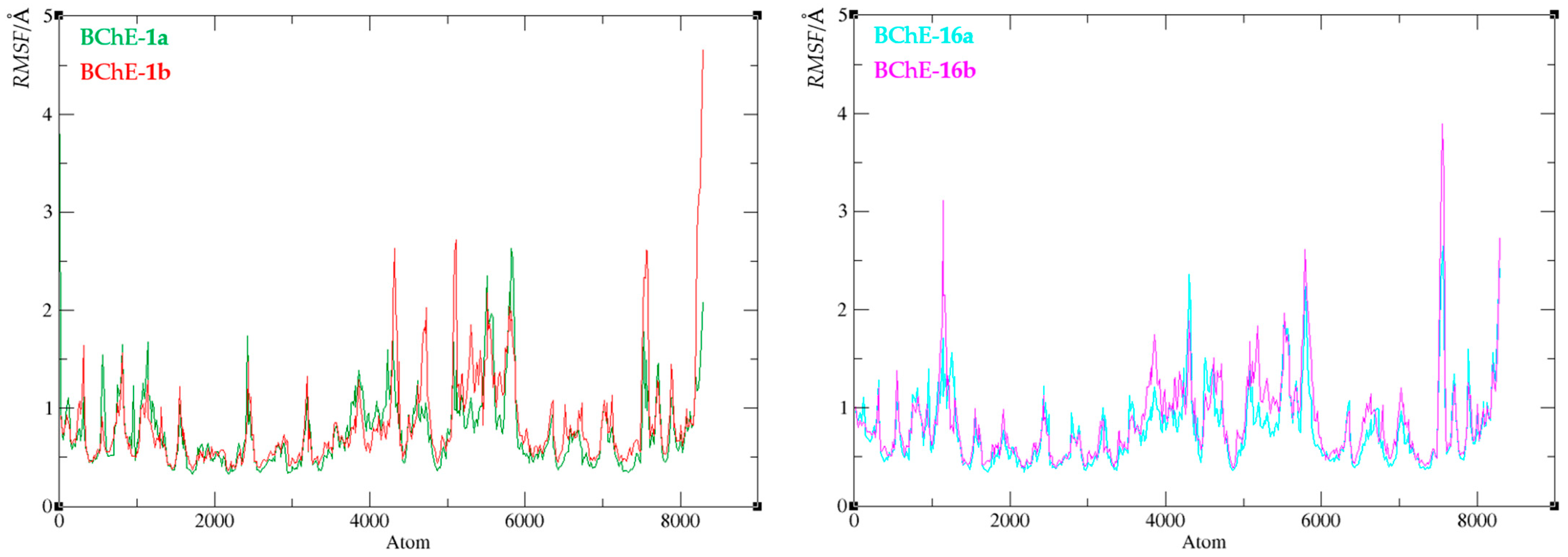
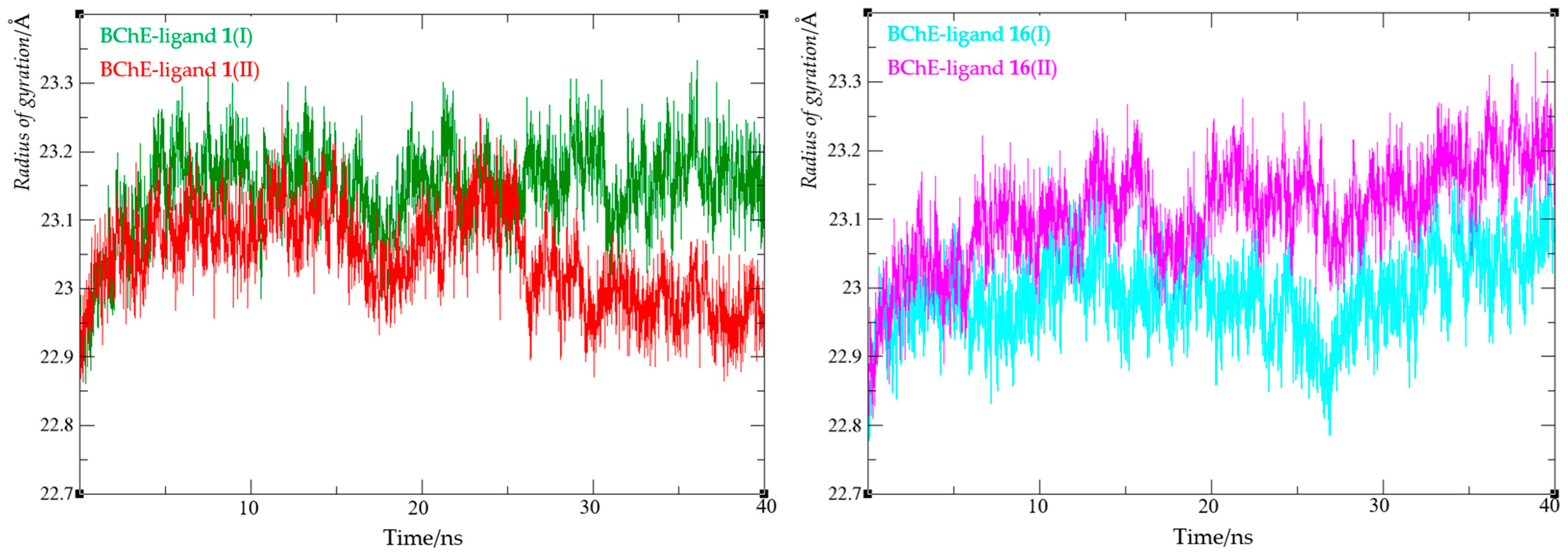
3.6. Computational Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. A Structural Model of Butyrylcholinesterase. Proc. Natl. Acad. Sci. USA 1997, 94, 3398–3403. [Google Scholar]
- Santos, A.L.; Carvalho, M.M.; Silva, D.F.; Amado, F.; Oliveira, C.R.; Pereira, C.M. Dual Inhibition of Acetylcholinesterase and Butyrylcholinesterase by Phenserine Improves Cognitive Function in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 16, 767–774. [Google Scholar] [CrossRef]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Hashimoto, Y. The Role of Butyrylcholinesterase in Alzheimer’s Disease and Its Potential as a Therapeutic Target. Neurobiol. Aging 2001, 22, 471–478. [Google Scholar] [CrossRef]
- Caccamo, A.; Oddo, S.; Sugarman, M.C.; LaFerla, F.M. Molecular Targets for Alzheimer’s Disease Therapy: Amyloid-Beta Aggregation and Cholinergic Dysfunction. Neurobiol. Aging 2010, 31, 987–999. [Google Scholar] [CrossRef]
- Vila, J.L.; Medina, M.; Correa, J.; Romero, L. Butyrylcholinesterase Inhibitors as New Drugs for Alzheimer’s Disease. J. Med. Chem. 2001, 44, 2022–2027. [Google Scholar] [CrossRef]
- Pereira, R.M.; Marques, L.A.; Silva, D.F.; Oliveira, C.R.; Pereira, C.M. Phenserine: A Selective Butyrylcholinesterase Inhibitor. Curr. Alzheimer Res. 2002, 4, 1–11. [Google Scholar]
- Röcken, C.; Bartels, M.; Jessberger, S.; Stühmer, W.; Alzheimer, C. The Pharmacological Profile of Rivastigmine as a Selective Inhibitor of Acetylcholinesterase and Butyrylcholinesterase. Neurochem. Int. 2003, 43, 315–323. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterase Inhibitors: From the Bench to the Bedside. Neurochem. Int. 2004, 45, 1–10. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Jakobsen, J.; Høgh-Rasmussen, E.; Holm, S.; Paulson, O.B. The Role of Acetylcholine and Acetylcholinesterase Inhibitors in Parkinson’s Disease. J. Clin. Neurosci. 2007, 14, 610–615. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A Review on Cholinesterase Inhibitors. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, Synthesis, and Biological Evaluation of Conformationally Restricted Rivastigmine Analogues. J. Med. Chem. 2004, 47, 5945–5952. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; et al. Novel Dual Inhibitors of AChE and MAO Derived from Hydroxy Aminoindan and Phenethylamine as Potential Treatment for Alzheimer’s Disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Ucar, G.; Gokhan, N.; Yesilada, A.; Bilgin, A.A. 1-N-Substituted Thiocarbamoyl-3-Phenyl-5-Thienyl-2-Pyrazolines: Novel Cholinesterase and Selective Monoamine Oxidase B Inhibitors for the Treatment of Parkinson’s and Alzheimer’s Diseases. Neurosci. Lett. 2005, 38, 327–331. [Google Scholar] [CrossRef]
- Toda, N.; Tago, K.; Marumoto, S.; Takami, K.; Ori, M.; Yamada, N.; Koyama, K.; Naruto, S.; Abe, K.; Yamazaki, R.; et al. A Conformational Restriction Approach to the Development of Dual Inhibitors of Acetylcholinesterase and Serotonin Transporter. Bioorganic Med. Chem. 2003, 11, 4389–4415. [Google Scholar] [CrossRef]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Švarcová, M.; Vinšová, J. Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules 2016, 21, 191. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Wang, Y.; Li, X.; Wang, S.; Wang, Z. Recent Advances on Carbamate-Based Cholinesterase Inhibitors as Potential Multifunctional Agents Against Alzheimer’s Disease. Eur. J. Med. Chem. 2022, 240, 114606. [Google Scholar] [CrossRef]
- Yanovsky, I.; Finkin-Groner, E.; Zaikin, A.; Lerman, L.; Shalom, H.; Zeeli, S.; Weill, T.; Ginsburg, I.; Nudelman, A.; Weinstock, M. Carbamate Derivatives of Indolines as Cholinesterase Inhibitors and Antioxidants for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2012, 55, 10700–10715. [Google Scholar] [CrossRef] [PubMed]
- Sviben, M.; Odak, I.; Barić, D.; Mlakić, M.; Horváth, O.; Fodor, L.; Roca, S.; Šagud, I.; Škorić, I. Resveratrol-Based Carbamates as Selective Butyrylcholinesterase Inhibitors: Design, Synthesis, Computational Study and Biometal Complexation Capability. Molecules 2025, 30, 316. [Google Scholar] [CrossRef]
- Mlakić, M.; Odak, I.; Barić, D.; Talić, S.; Šagud, I.; Štefanić, Z.; Molčanov, K.; Lasić, Z.; Kovačević, B.; Škorić, I. New Resveratrol Analogs as Improved Biologically Active Structures: Design, Synthesis and Computational Modeling. Bioorganic Chem. 2024, 143, 106965. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrysAlis PRO; Rigaku Corporation: Wrocław, Poland, 2024. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Faraho, I.; Odak, I.; Kovačević, B.; Raspudić, A.; Šagud, I.; Bosnar, M.; Škorić, I.; Barić, D. Cholinesterase Inhibitory and Anti-Inflammatory Activity of the Naphtho- and Thienobenzo-Triazole Photoproducts: Experimental and Computational Study. Int. J. Mol. Sci. 2023, 24, 14676. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Colletier, J.P. Crystal Structure of Human Butyrylcholinesterase in Complex with 2-{1-[4-(12-Amino-3-chloro-6,7,10,11-tetrahydro-7,11-methanocycloocta[b]quinolin-9-yl)butyl]-1H-1,2,3-triazol-4-yl}-N-[4-hydroxy-3-methoxybenzyl]acetamide. PDB 7AIY. Available online: https://www.wwpdb.org/pdb?id=pdb_00007aiy (accessed on 25 March 2021).
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Mencaroni, L.; Carlotti, B.; Cesaretti, A.; Elisei, F.; Grgičević, A.; Škorić, I.; Spalletti, A. Competition between fluorescence and triplet production ruled by nitro groups in one-arm and two-arm styrylbenzene heteroanalogues. Photochem. Photobiol. Sci. 2020, 19, 1665–1676. [Google Scholar] [CrossRef]
- Carlotti, B.; Cesaretti, A.; Cacioppa, G.; Elisei, F.; Odak, I.; Škorić, I.; Spalletti, A. Fluorosolvatochromism and hyperpolarizability of one-arm and two-arms nitro compounds bearing heterocyclic rings. J. Photochem. Photobiol. A Chem. 2019, 368, 190–199. [Google Scholar] [CrossRef]
- Kikaš, I.; Škorić, I.; Marinić, Ž.; Šindler-Kulyk, M. Synthesis and Phototransformations of Novel Styryl-substituted Furobenzobicyclo[3.2.1]octadiene Derivatives. Tetrahedron 2010, 66, 9405–9414. [Google Scholar] [CrossRef]
- Škorić, I.; Šmehil, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Photochemistry of ω-(o-vinylphenyl)-ω’-(phenyl/2-furyl) butadienes: New approach to 4-substituted benzobicyclo[3.2.1]octadienes. J. Photochem. Photobiol. A Chemistry 2009, 207, 190–196. [Google Scholar] [CrossRef]
- Hasselgren, C.; Bercu, J.; Cayley, A.; Cross, K.; Glowienke, S.; Kruhlak, N.; Muster, W.; Nicolette, J.; Vijayaraj Reddy, M.; Saiakhov, R.; et al. Management of Pharmaceutical ICH M7 (Q)SAR Predictions–The Impact of Model Updates. Regul. Toxicol. Pharmacol. 2020, 118, 104807. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development, 2nd ed.; Faqi, A.S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 39–67. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- AI-DrugLab, Singapore Management University. Available online: https://ai-druglab.smu.edu/admet (accessed on 15 April 2025).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 4 |
|---|---|---|
| Empirical formula | C14H13NO2S | C15H15NO2S |
| Formula wt./g mol−1 | 259.31 | 273.34 |
| Crystal dimensions/mm | 0.3 × 0.03 × 0.02 | 0.1 × 0.05 × 0.02 |
| Space group | P21 | P21/c |
| a/Å | 5.07910(10) | 5.12806(17) |
| b/Å | 16.6438(2) | 30.1746(7) |
| c/Å | 15.4486(2) | 9.2923(4) |
| α/° | 90 | 90 |
| β/° | 98.0250(10) | 105.137(4) |
| γ/° | 90 | 90 |
| Z | 4 | 4 |
| V/Å3 | 1293.17(3) | 1387.97(8) |
| Dcalc/g cm−3 | 1.332 | 1.308 |
| μ/mm−1 | 2.171 | 2.049 |
| Θ range/° | 3.93–80.03 | 5.12–75.85 |
| T(K) | 104(6) | 293(2) |
| Radiation wavelength | 1.54184 (CuKα) | 1.54184 (CuKα) |
| Diffractometer type | XtaLAB Synergy, Dualflex, HyPix | XtaLAB Synergy, Dualflex, HyPix |
| Range of h, k, l | −6 > h > 6; −21 > k > 21; −19 > l > 19 | −6 > h > 6; −26 > k > 37; −11 > l > 11 |
| Reflections collected | 17951 | 25937 |
| Independent reflections | 5485 | 2990 |
| Observed reflections (I ≥ 2σ) | 5146 | 2337 |
| Rint | 0.0776 | 0.0899 |
| R (F) | 0.0508 | 0.0743 |
| Rw (F2) | 0.1369 | 0.2130 |
| No. of parameters, restraints | 335, 1 | 181, 0 |
| Goodness of fit | 1.039 | 1.054 |
| Δρmax, Δρmin (eÅ−3) | 0.48; −0.64 | 0.66; −0.51 |
| D–H/Å | H···A/Å | D···A/Å | D–H···A/º | Symm. op. on A | |
|---|---|---|---|---|---|
| Compound 1 | |||||
| N1–H1A∙∙∙O2 | 0.84 | 2.11 | 2.824(4) | 143 | −1 + x, y, z |
| N2–H2A∙∙∙O4 | 0.93 | 2.10 | 2.821(4) | 134 | 1 + x, y, z |
| C6–H6∙∙∙S1 | 0.95 | 2.73 | 3.156(5) | 108 | x, y, z |
| C6–H6∙∙∙O1 | 0.95 | 2.46 | 2.828(5) | 103 | x, y, z |
| C10–H10∙∙∙O4 | 0.95 | 2.57 | 3.477(5) | 160 | 1 + x, y, 1 + z |
| C16–H16∙∙∙O2 | 0.95 | 2.60 | 3.397(5) | 142 | x, y, z |
| C20–H20∙∙∙S2 | 0.95 | 2.73 | 3.158(3) | 108 | x, y, z |
| C20–H20∙∙∙O3 | 0.95 | 2.46 | 2.821(4) | 103 | x, y, z |
| Compound 4 | |||||
| N1–H1A∙∙∙O2 | 0.86 | 2.14 | 2.892(3) | 145.4 | −1 + x, y, z |
| C6–H6∙∙∙S1 | 0.93 | 2.83 | 3.210(4) | 106 | x, y, z |
| C6–H6∙∙∙O1 | 0.93 | 2.45 | 2.800(4) | 103 | x, y, z |
| π···π | Cg a···Cg/ Å | α b/º | β c/º | Cg···Plane(Cg2)/Å | Offset/ Å | Symm. |
|---|---|---|---|---|---|---|
| Compound 1 | ||||||
| S1→C4∙∙∙S1→C4 | 5.079(3) | 0.0(3) | 50.0 | 3.264(2) | 3.891 | −1 + x, y, z |
| S1→C4∙∙∙S2→C18 | 4.246(3) | 6.1(2) | 33.7 | 3.751(2) | 2.355 | x, y, z |
| C7→C12∙∙∙C7→C12 | 5.079(2) | 0.0(17) | 50.4 | 3.2388(15) | 3.912 | −1 + x, y, z |
| C7→C12∙∙∙C21→C26 | 5.521(2) | 0.78(17) | 58.4 | 2.8750(15) | 4.703 | 1 + x, y, z |
| S2→C18∙∙∙S1→C4 | 4.246(3) | 6.1(2) | 27.9 | 3.5336(19) | 1.989 | x, y, z |
| S2→C18∙∙∙S2→C18 | 5.079(3) | 0.0(2) | 52.6 | 3.0846(19) | 4.035 | −1 + x, y, z |
| C21→C26∙∙∙C7→C12 | 5.521(2) | 0.78(17) | 58.6 | 2.8916(15) | 4.713 | −1 + x, y, −1 + z |
| C21→C26∙∙∙C21→C26 | 5.079(2) | 0.03(17) | 49.6 | 3.2914(15) | 3.868 | −1 + x, y, z |
| Compound 4 | ||||||
| S1→C4∙∙∙S1→C4 | 5.128(2) | 0.00(19) | 54.6 | 2.9734(16) | 4.178 | −1 + x, y, z |
| S1→C4∙∙∙C7→C12 | 5.269(2) | 8.71(18) | 41.0 | 3.4207(16) | 3.458 | −1 + x, y, −1 + z |
| C7→C12∙∙∙C7→C12 | 5.128(2) | 0.00(16) | 46.1 | 3.5526(14) | 3.698 | −1 + x, y, z |
| C–H···π | H···Cg/ Å | γ a/º | C–H···Cg/Å | C···Cg/Å | Symm. Operation on Cg |
|---|---|---|---|---|---|
| C15–H15A∙∙∙C7→C12 | 2.68 | 11.24 | 171 | 3.627(4) | 1 + x, y, z |
| Compound | AChE | BChE | ||
|---|---|---|---|---|
| % Inhibition * | IC50/μM | % Inhibition * | IC50/μM | |
| 1 | 36.82 ± 0.20 (250) | – | 84.37 ± 1.18 (10) | 0.500 |
| 2 | 30.07 ± 2.38 (100) | – | 84.66 ± 3.20 (100) | 3.740 |
| 3 | 19.01 ± 1.10 (500) | – | 82.15 ± 2.00 (500) | 149.7 |
| 4 | 15.43 ± 0.30 (50) | – | 85.38 ± 2.29 (10) | 0.913 |
| 5 | 34.60 ± 1.39 (100) | – | 74.79 ± 2.36 (5) | 1.102 |
| 6 | 19.80 ± 0,65 (50) | – | 74.00 ± 3.20 (50) | 14.053 |
| 7 | 50.05 ± 1.10 (100) | – | 69.80 ± 1.15 (100) | 30.403 |
| 8 | 87.21 ± 0.47 (500) | 275.5 | 87.21 ± 0.47 (500) | 59.108 |
| 9 | 28.22 ± 1.06 (100) | – | 83.91 ± 1.77 (10) | 0.583 |
| 10 | 23.33 ± 1.67 (68) | – | 83.48 ± 1.49 (27) | 3.081 |
| 11 | 20.71 ± 0.30 (50) | – | 82.75 ± 1.57 (10) | 1.541 |
| 12 | 17.85 ± 0.51 (100) | – | 83.19 ± 0.84 (50) | 1.067 |
| 13 | 29.87 ± 0.31 (50) | – | 84.41 ± 2.29 (25) | 1.883 |
| 14 | 20.92 ± 0.46 (250) | – | 70.00 ± 0.80 (250) | 90.001 |
| 16 | 23.44 ± 1.57 (100) | – | 84.25 ± 0.82 (1) | 0.0265 |
| 17 | 16.13 ± 1.24 (100) | – | 82.28 ± 2.44 (25) | 0.645 |
| 18 | 22.20 ± 1.17 (25) | – | 87.48 ± 1.31 (25) | 0.430 |
| 19 | 41.35 ± 0.92 (50) | – | 79.67 ± 2.51 (50) | 2.045 |
| Galantamine | 90.5 ± 1.3 (60) | 0.15 | 90.10 ± 3.40 (4.5) | 7.90 |
| Structure | ICH M7 Class | Cohort of Concern | Derek Prediction | Sarah Prediction | Experimental Data | Overall in Silico |
|---|---|---|---|---|---|---|
| 1 | Class 3 | No |  |  | Carc: Unspecified Ames: Unspecified | Positive |
| 2 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 3 | Class 5 | No | |  | Carc: Unspecified Ames: Unspecified | Negative |
| 4 | Class 5 | No | |  | Carc: Unspecified Ames: Unspecified | Negative |
| 5 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 6 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 7 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 8 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 9 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 10 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 11 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| 12 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 13 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 14 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 15 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 16 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 17 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 18 | Class 5 | No | | | Carc: Unspecified Ames: Unspecified | Negative |
| 19 | Class 3 | No | | | Carc: Unspecified Ames: Unspecified | Positive |
| Property | Model Name | 9 | 16 | 18 | Bambuterol | Rivastigmine | Unit |
|---|---|---|---|---|---|---|---|
| Absorption | Water solubility | −4.358 | −4.272 | −4.547 | log mol/L | ||
| Caco2 | 1.907 | 2.117 | 1.962 | log Papp in 10−6 cm/s | |||
| Intestinal absorption | 90.33 | 91.775 | 92.085 | % Absorbed | |||
| Skin permeability | −2.693 | −2.743 | −2.757 | log Kp | |||
| P−glycoprotein substrate | No | No | Yes | ||||
| P−glycoprotein I inhibitor | No | Yes | Yes | ||||
| P−glycoprotein II inhibitor | No | No | No | ||||
| Distribution | VDss (human) | 0.4 | 0.298 | 0.324 | 0.401 | 0.451 | log L/kg |
| Fraction unbound | 0.022 | 0.033 | 0.025 | − | − | Fu | |
| BBB permeability | 0.343 | 0.315 | 0.261 | 0.707 | 0.968 | log BB | |
| CNS permeability | −0.979 | −1.069 | −1.063 | −1.95 | −0.801 | log PS | |
| Metabolism | CYP2D6 substrate | No | No | No | |||
| CYP3A4 substrate | Yes | Yes | Yes | ||||
| CYP1A2 inhibitor | Yes | Yes | Yes | ||||
| CYP2C19 inhibitor | Yes | Yes | Yes | ||||
| CYP2C9 inhibitor | Yes | No | Yes | ||||
| CYP2D6 inhibitor | No | No | No | ||||
| CYP3A4 inhibitor | No | No | No | ||||
| Excretion | Total clearance | 0.138 | 0.373 | 0.262 | log ml/min/kg | ||
| Renal OCT2 substrate | Yes | No | No | Yes/no | |||
| Toxicity | AMES toxicity | No | No | No | Yes/no | ||
| Max. tolerated dose | 0.292 | −0.157 | −0.249 | log mg/kg/day | |||
| hERG I inhibitor | No | No | No | ||||
| hERG II inhibitor | Yes | Yes | Yes | ||||
| Oral rat acute toxicity (LD50) | 2.648 | 2.618 | 2.656 | mol/kg | |||
| Oral rat chronic toxicity (LOAEL) | 1.896 | 1.281 | 1.34 | log mg/kg_bw/day | |||
| Hepatotoxicity | Yes | No | No | ||||
| Skin sensitization | No | No | No | ||||
| T. pyriformis toxicity | 1.812 | 1.465 | 1.542 | log ug/L | |||
| Minnow toxicity | 0.214 | −0.487 | −0.62 | log mM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raspudić, A.; Odak, I.; Mlakić, M.; Jelčić, A.; Bulava, K.; Karadža, K.; Milašinović, V.; Šagud, I.; Pongrac, P.; Štefok, D.; et al. Heterostilbene Carbamates with Selective and Remarkable Butyrylcholinesterase Inhibition: Computational Study and Physico-Chemical Properties. Biomolecules 2025, 15, 825. https://doi.org/10.3390/biom15060825
Raspudić A, Odak I, Mlakić M, Jelčić A, Bulava K, Karadža K, Milašinović V, Šagud I, Pongrac P, Štefok D, et al. Heterostilbene Carbamates with Selective and Remarkable Butyrylcholinesterase Inhibition: Computational Study and Physico-Chemical Properties. Biomolecules. 2025; 15(6):825. https://doi.org/10.3390/biom15060825
Chicago/Turabian StyleRaspudić, Anamarija, Ilijana Odak, Milena Mlakić, Antonija Jelčić, Karla Bulava, Karla Karadža, Valentina Milašinović, Ivana Šagud, Paula Pongrac, Dora Štefok, and et al. 2025. "Heterostilbene Carbamates with Selective and Remarkable Butyrylcholinesterase Inhibition: Computational Study and Physico-Chemical Properties" Biomolecules 15, no. 6: 825. https://doi.org/10.3390/biom15060825
APA StyleRaspudić, A., Odak, I., Mlakić, M., Jelčić, A., Bulava, K., Karadža, K., Milašinović, V., Šagud, I., Pongrac, P., Štefok, D., Barić, D., & Škorić, I. (2025). Heterostilbene Carbamates with Selective and Remarkable Butyrylcholinesterase Inhibition: Computational Study and Physico-Chemical Properties. Biomolecules, 15(6), 825. https://doi.org/10.3390/biom15060825