1. Introduction
Eukaryotic cells have evolved complex molecular machineries to protect genome integrity, including intricate DNA repair pathways and mechanisms ensuring accurate chromosome segregation during cell division. Central to these processes are the Structural Maintenance of Chromosomes (SMC) protein complexes, which play crucial roles in chromosome organization, condensation, cohesion, and DNA repair [
1]. Among these, the SMC-5/6 complex is uniquely implicated in resolving complex DNA structures that arise during DNA replication, recombination, and repair, thereby safeguarding against genomic instability [
2]. The SMC-5/6 complex is conserved across eukaryotes and minimally comprises the core SMC-5 and SMC-6 proteins, which heterodimerize via their hinge domains and associate through their ATPase head domains, bridged by the kleisin subunit NSE-4 (Non-SMC Element 4). Additional subunits, including NSE-1, NSE-2 (a SUMO ligase), and NSE-3, associate with this core to form the functional holocomplex [
3,
4,
5]. NSE-1, an essential kleisin-interacting protein with ubiquitin E3 ligase activity potential, is crucial for complex stability and function, often serving as a marker for the complex’s chromosomal localization [
6]. Studies in various organisms have demonstrated that dysfunction of the SMC-5/6 complex leads to hypersensitivity to DNA damaging agents, replication stress, defects in homologous recombination repair, chromosome segregation errors, and, in humans, developmental disorders and predisposition to cancer [
7,
8,
9]. Despite its established importance, the precise molecular mechanisms by which SMC-5/6 subunits coordinate their activities, how the complex is recruited to specific chromosomal sites, and how its functions are regulated remain areas of active investigation. In particular, understanding the specific contributions of individual domains and residues within core components like SMC-5 to complex assembly, localization, and functional output requires detailed structure–function analysis. While null mutations demonstrate the essential nature of the complex, they often preclude the study of specific functional aspects that might be retained in separation-of-function or hypomorphic alleles.
The nematode
Caenorhabditis elegans offers a powerful system to dissect the functions of conserved genome maintenance factors like the SMC-5/6 complex. Our previous work using fluorescently tagged subunits, such as NSE-1::GFP, has visualized the dynamic localization of the complex to chromosomes, particularly in the germline where meiotic recombination and DNA repair are prominent [
10,
11]. Perturbations leading to mislocalization of tagged subunits often correlate with functional deficits [
10,
11]. To gain deeper insights into the regulation of SMC-5/6 function and localization, we utilized a
C. elegans strain expressing NSE-1 tagged with GFP,
nse-1::gfp(wsh1), as a reporter for complex integrity and chromosomal association. We performed a forward genetic screen using ethyl methanesulfonate (EMS) mutagenesis to identify mutations causing aberrant NSE-1::GFP distribution in the germline. Here, we report the isolation and characterization of three novel mutant alleles (
smc-5(wsh31),
smc-5(wsh32), and
smc-5(wsh33)) exhibiting distinct NSE-1::GFP mislocalization patterns. Through genetic mapping and whole-genome sequencing, we identified causative mutations within the
smc-5 gene, encoding a core component of the complex. These alleles, comprising two truncations (
smc-5(wsh31) and
smc-5(wsh32)) and a missense mutation in a highly conserved residue (
smc-5(wsh33), Y975D), provide a valuable allelic series. We performed functional analysis, assessing their impact on fertility, progeny viability, chromosome segregation fidelity, and sensitivity to various genotoxic agents (MMS, HU, and Cisplatin) at different developmental stages. Our findings reveal differential requirements for SMC-5 domains and specific residues in maintaining complex integrity, ensuring proper NSE-1 localization, and executing critical DNA repair functions, particularly under conditions of replication stress and interstrand crosslink damage. Our study thus fills a critical knowledge gap by linking a defined SMC-5 domain to the regulation of SMC-5/6 complex assembly and function in vivo.
2. Materials and Methods
2.1. EMS Mutagenesis of nse-1::gfp(wsh1) Worms
nse-1::gfp(wsh1) worms were grown on NGM agar plates seeded with E. coli OP50 until most animals reached the L4 stage, at which point they were washed off with M9 buffer into a 15 mL conical tube, briefly centrifuged, and washed once more to remove debris. The final worm pellet was resuspended in 2 mL M9, and in a fume hood a 0.05 M solution of ethyl methanesulfonate (EMS, Sigma #M-0880, Merck KGaA, Darmstadt, Germany) was prepared by adding 10 µL of liquid EMS to 2 mL of M9 in a separate 15 mL tube and gently mixing until the dense liquid dissolved. This EMS solution was combined with the 2 mL worm suspension to yield a final concentration of 0.025 M EMS, sealed with Parafilm, and placed on a rocker at 20 °C for 4 h. All handling and mixing steps were performed using disposable plasticware in a fume hood, and EMS-contaminated items (pipettes, tubes, and residual solutions) were inactivated for 24 h by mixing with an equal volume of 0.1 M NaOH and 20% (w/v) sodium thiosulfate. After the incubation period, the worms were washed twice with M9, transferred with a Pasteur pipette to the edge of an OP50 lawn (avoiding the use of plastic pipette tips to prevent the worms from sticking), and allowed to recover for 15–20 min, at which point the healthy late-L4 worms were picked as P0 animals. Post-mutagenesis, animals were washed four times in M9 and dispensed onto OP50 plates, where they were grown for two days to lay eggs; the resulting L1 larvae were then filtered as the F1 generation, frozen as a library, and also plated again on OP50 for another two days, after which the next batch of L1 larvae were filtered and designated F2, then similarly frozen as a library.
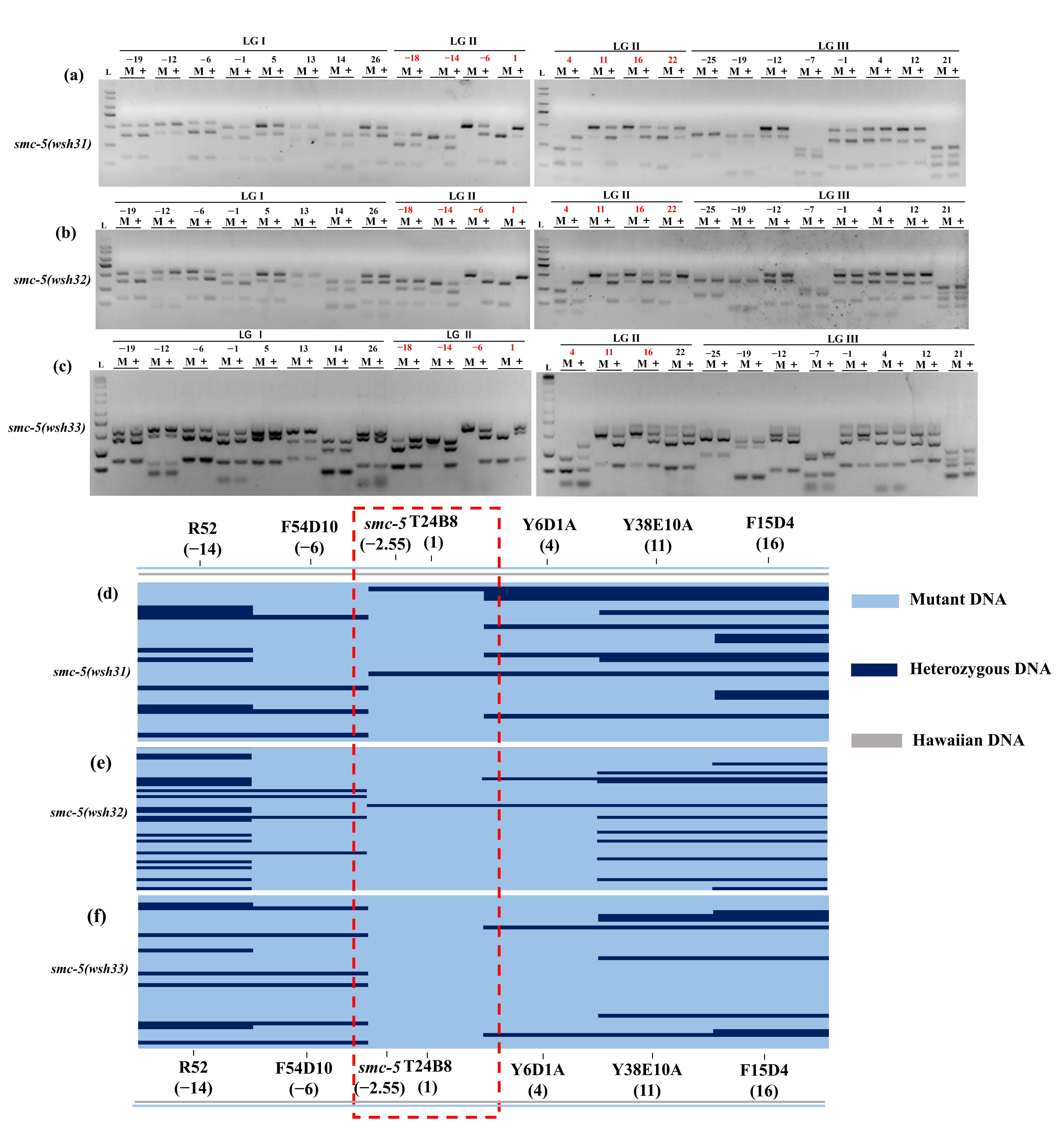
2.2. SNP-Based Mapping Protocol for the Identification of Mutations in the Mutant Strains Isolated
We crossed EMS-induced mutants in the
nse-1::gfp(wsh1) background to CB4856 (Hawaiian) males and isolated F1 heterozygotes, allowing them to self-fertilize to produce F2 progeny from which homozygous mutant and non-mutant pools were prepared to facilitate “bulk segregant” chromosome mapping, as described by Davis et al. (2005) [
12]. For each pool, 30–50 animals were lysed in 20 µL of single-worm lysis buffer (10 mM Tris-HCl pH 8.3, 50 mM KCl, 2.5 mM MgCl
2, 0.45% NP-40 or IGEPAL CA-630, 0.45% Tween-20, 0.01% gelatin, and 60 µg/mL Proteinase K) by incubation at 65 °C for 1 h followed by 95 °C for 15 min. PCR master mixes were assembled for each SNP marker, with Taq polymerase, dNTPs, and a uniform annealing temperature of 60 °C, and were arrayed into 96-well plates such that mutant and non-mutant lysates could be dispensed into alternate rows. Primers designed to flank DraI-based SNPs at evenly spaced intervals across all six chromosomes were pin-replicated into the master mixes, and after amplification, the PCR products were digested with DraI in the same plate. Samples were analyzed on 2.5% agarose gels, and enrichment of the Bristol allele in the mutant lane was used to infer linkage. Once the candidate chromosome was identified, individual homozygous F2 recombinants were singled and allowed to produce F3 progeny; these populations were each lysed separately, and their templates were again analyzed by PCR and DraI digestion for multiple markers spanning the linked interval. Genotype transitions (from Bristol to Hawaiian or vice versa) among the recombinants pinpointed the region harboring each mutation, enabling fine-scale interval mapping according to the streamlined protocol of Davis et al. [
12].
2.3. Phenotypic Assessment
To evaluate brood size, 25 L4-stage hermaphrodite worms from each strain were individually placed on NGM plates seeded with OP50 bacteria. Every 12 h, each worm was transferred to a fresh plate, and the number of eggs laid on the previous plate was recorded. This process continued until egg-laying ceased, allowing for the calculation of the total number of fertilized eggs per hermaphrodite. To assess progeny viability, the number of unhatched eggs was counted 24 h post-laying, and viability was expressed as the percentage of hatched larvae relative to the total number of eggs laid. Additionally, 72 h after egg-laying commenced, male progeny were identified and counted to determine the percentage of male offspring.
2.4. Genotoxic Assays
To assess the impact of genotoxic agents, synchronized populations of at least 50
C. elegans larvae at either the L1 or L4 stage were exposed to varying concentrations of methyl methanesulfonate (MMS), cisplatin, or hydroxyurea (HU), following established protocols [
13,
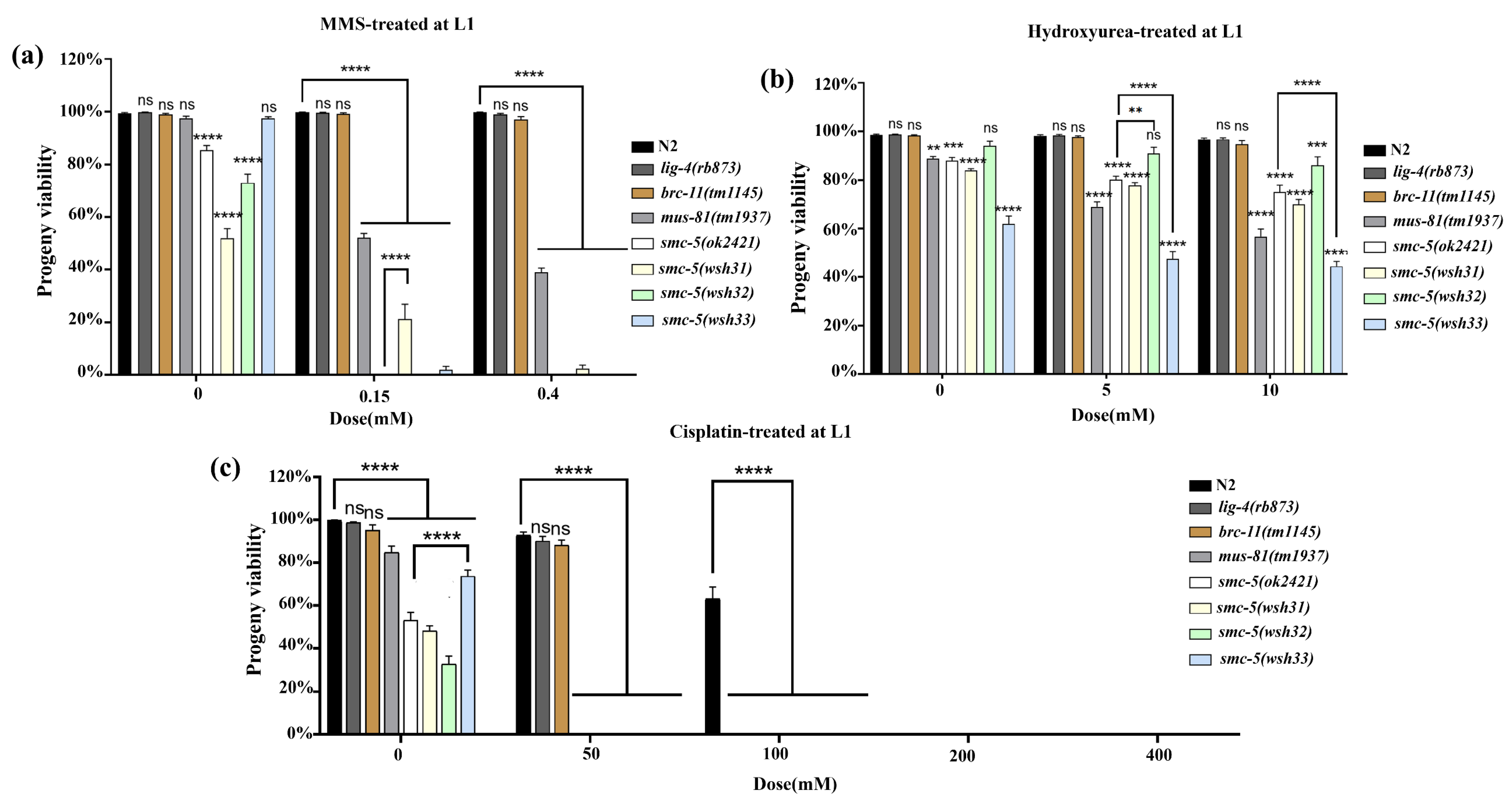
14]. For MMS and cisplatin treatments, worms were incubated with the agents at specified doses for 16 h; HU exposure was conducted over a 20 h period. Post-treatment, L1 larvae were allowed a recovery period of 24 h, while L4 larvae recovered for 24 h. For the L1 progeny viability assay, treated larvae were allowed to recover for 72 h (day 3 post-treatment). By this point, individuals capable of maturing had reached the adult stage, whereas developmentally arrested animals were mostly immobile or dead and could be readily excluded. Only the surviving adults were transferred to brood plates for viability scoring. After recovery, five adult worms from each group were individually placed on NGM plates seeded with OP50 bacteria to lay eggs over a 6–8 h period. Parent worms were then removed, and the number of eggs laid was recorded. After an additional 24 h, unhatched eggs were counted to determine progeny viability, calculated as the percentage of hatched larvae relative to the total number of eggs laid. Each experiment was performed in triplicate and repeated independently three times to ensure reproducibility.
2.5. Developmental Assays Following Genotoxic Treatment
To assess the impact of DNA-damaging agents on
C. elegans development, synchronized L1 larvae were obtained by filtering through an 11 μm nylon mesh (Millipore, Burlington, MA, USA) using M9 buffer. These larvae were then exposed to varying concentrations of methyl methanesulfonate (MMS) and cisplatin for 16 h, or hydroxyurea (HU) for 20 h, with specific dosages detailed in the Results section [
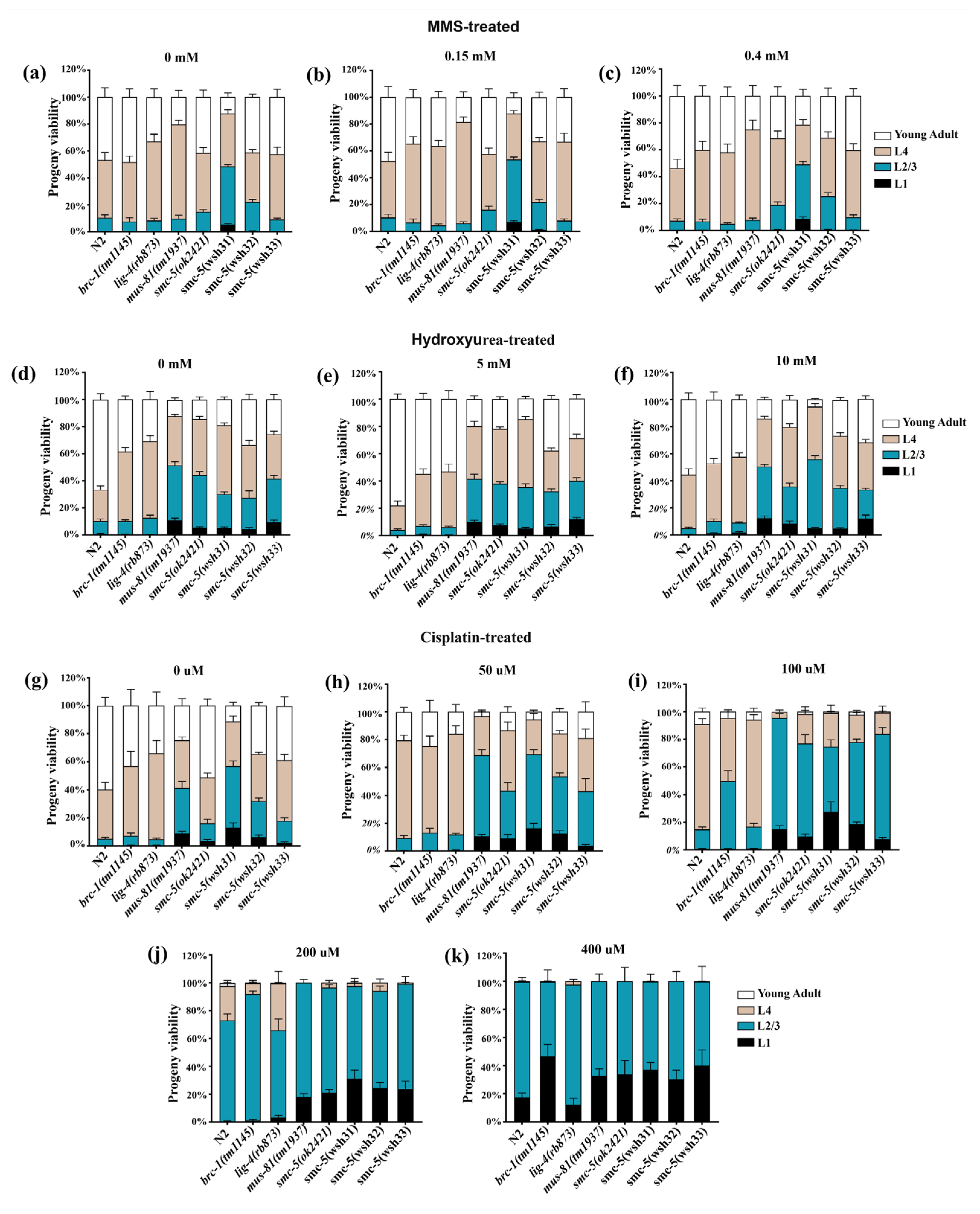
10]. Post-treatment, the larvae were thoroughly rinsed with M9 buffer to remove residual agents, evenly distributed onto NGM agar plates seeded with OP50 bacteria, and allowed to recover at 20 °C for 48 h. Subsequently, their developmental stages were evaluated under a stereomicroscope. To ensure the reliability and reproducibility of the findings, each experiment was independently conducted twice.
2.6. Yeast Two-Hybrid Assay for Interaction Between NSE-4 and SMC-5 (Y975D) Mutant
Cultures of Y2HGold yeast cells were first transformed by the lithium acetate method with pairs of plasmids encoding bait (pGBKT7 constructs) and prey (pGADT7 constructs) for NSE-4 or smc-5 (including the smc-5 Y975D mutant), using a mixture containing approximately 2.5 µL of each plasmid, 240 µL of 50% PEG3350, 36 µL of 1 M LiAc, and 10 µL of salmon sperm DNA (10 mg/mL). Transformants were spread on synthetic dropout (SD) medium lacking tryptophan and leucine (SD-Trp-Leu) to select for both plasmids, incubated at 30 °C for three days, and single colonies were restreaked to ensure purity. For strain activation and verification, overnight cultures grown in SD-Trp-Leu liquid medium at 30 °C and 200 rpm were adjusted to OD600 ~0.05, then 5 µL of each suspension was spotted onto plates lacking tryptophan and leucine (SD-Trp-Leu) as well as plates additionally lacking histidine (SD-Trp-Leu-His), with or without 3-amino-1,2,4-triazole (3-AT). Growth after 2–3 days at 30 °C was compared to determine whether the bait and prey constructs interacted sufficiently to activate reporter genes, as indicated by colony formation on the more stringent selection media.
2.7. Statistical Analysis
To assess the significance of differences within our datasets, we employed statistical tests including the one-way ANOVA with Fisher’s least significant difference (LSD) test, two-way ANOVA with Dunnett’s multiple comparisons test, and two-tailed chi-squared tests with Fisher’s exact test, as appropriate. The specific test applied for each comparison is detailed in the corresponding figure legends. Statistical significance was denoted as follows: p > 0.05 (ns) indicating no significant difference, p ≤ 0.05 indicating significance at the 5% level, p ≤ 0.01 at the 1% level, and p ≤ 0.0001 at the 0.01% level. For unpaired two-tailed Student’s t-tests, significance levels were set at p > 0.05 (ns) for no significant difference, p ≤ 0.05 for significance at the 5% level, and p ≤ 0.01 at the 1% level. Data are presented as means, with error bars representing the standard error of the mean (SEM).
4. Discussion
In this study, we employed a forward genetic screen in
C. elegans based on the localization of NSE-1::GFP to identify factors crucial for the function and chromosomal association of the SMC-5/6 complex. Our screen successfully isolated three independent mutants,
smc-5(wsh31),
smc-5(wsh32), and
smc-5(wsh33), all exhibiting aberrant NSE-1::GFP distribution in the germline. Subsequent genetic mapping and whole-genome sequencing pinpointed the causative lesions to the
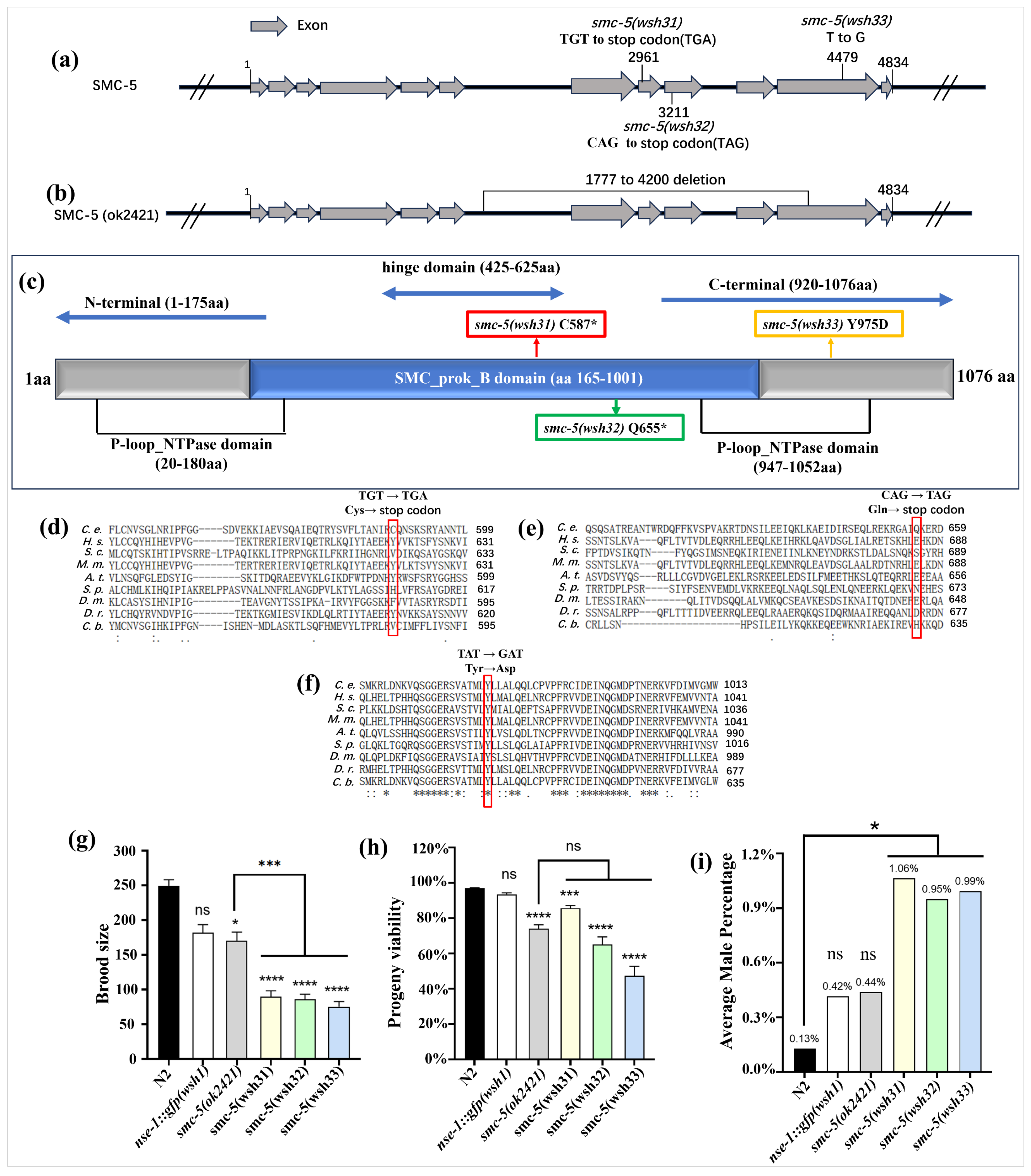
smc-5 gene, identifying three novel alleles:
smc-5(wsh31) (C587*),
smc-5(wsh32) (Q655*), and
smc-5(wsh33) (Y975D). These alleles provide a valuable resource for dissecting SMC-5 function, complementing the existing null allele
smc-5(ok2421) [
28]. A primary finding is the distinct impact of these
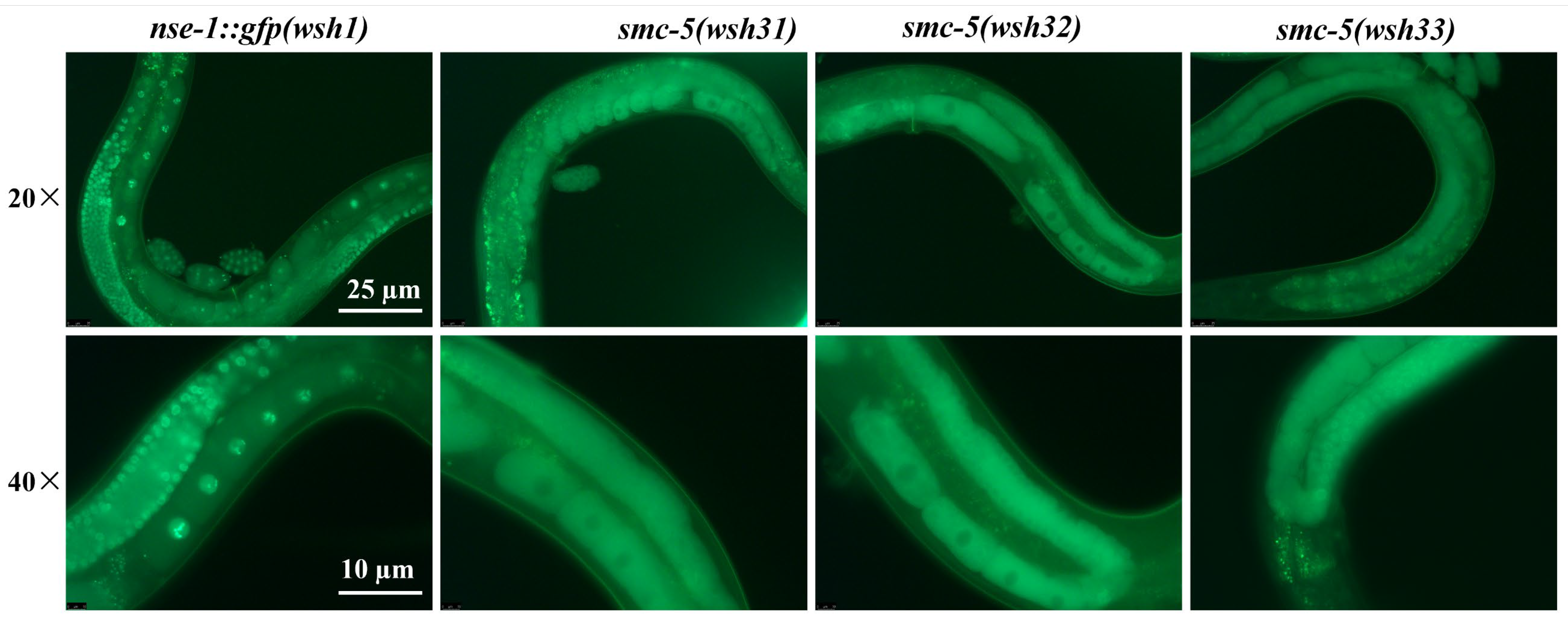
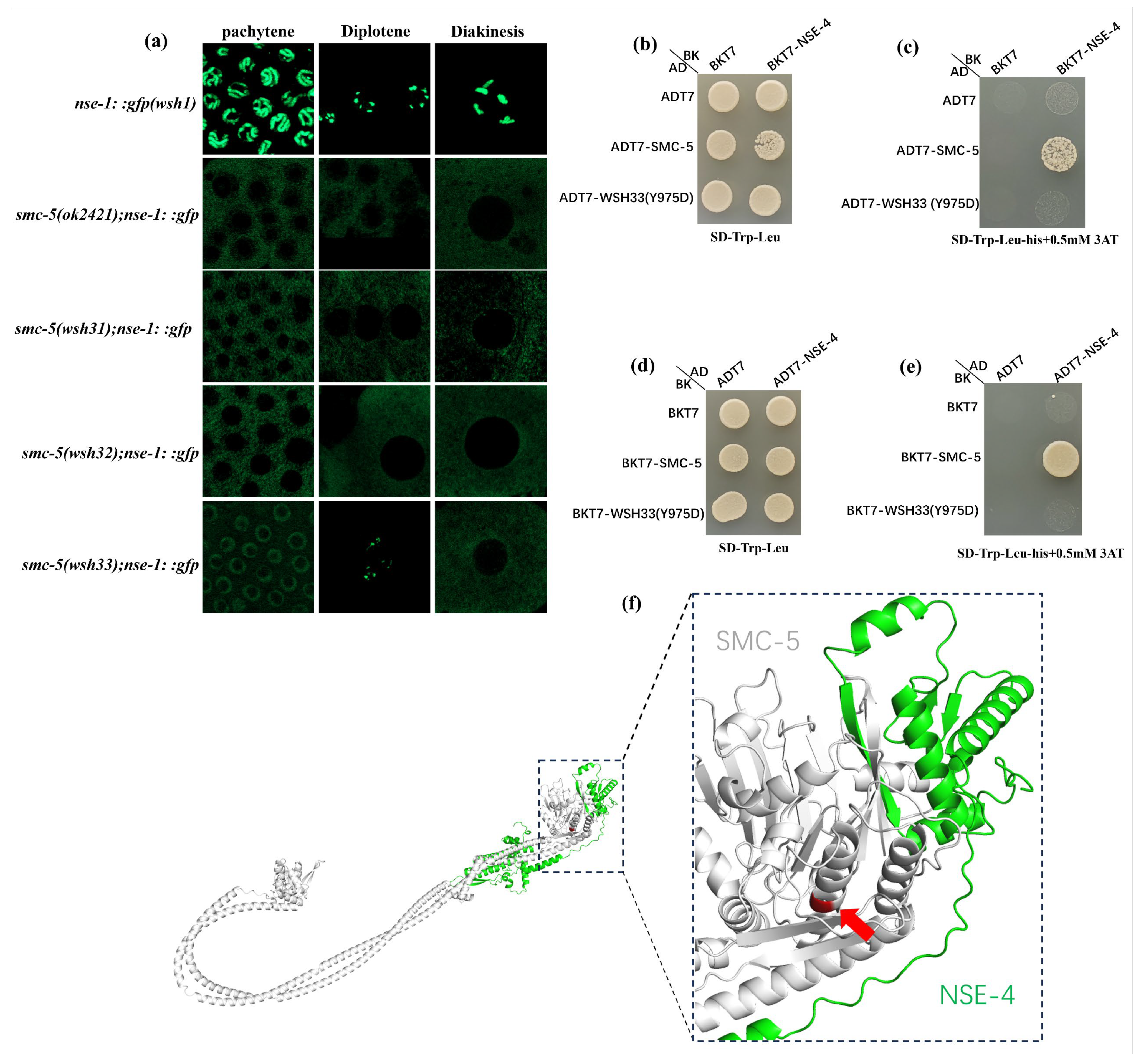
smc-5 alleles on the subcellular localization of NSE-1::GFP. The two truncating mutations,
smc-5(wsh31) and
smc-5(wsh32), resulted in a phenotype closely resembling the
smc-5(ok2421) null allele: NSE-1::GFP was excluded from the nucleus and failed to associate with meiotic chromosomes (
Figure 3 and
Figure 7a). This observation aligns with previous reports suggesting that the integrity of the core SMC-5/6 heterodimer is essential for the stable nuclear retention and chromatin loading of NSE subunits [
6,
15]. The premature stop codons in
wsh31 and
wsh32 likely lead to unstable or non-functional SMC-5 protein fragments incapable of proper complex assembly, resulting in the apparent cytoplasmic sequestration or degradation of NSE-1::GFP. Intriguingly, the missense allele
smc-5(wsh33) (Y975D) conferred a distinct, partial, and stage-dependent NSE-1::GFP localization defect (
Figure 3 and
Figure 7a). While NSE-1::GFP was substantially delocalized from chromosomes at pachytene (though less severely than in truncation/null mutants), it exhibited partial re-association during diplotene, only to become mislocalized again by diakinesis. This dynamic and incomplete mislocalization suggests that the Y975D mutation compromises, but does not abolish, SMC-5 function related to NSE-1 recruitment or retention [
18,
19]. The Y975 residue is located within the C-terminal portion of the SMC domain, near the ATPase head region. Its striking conservation across diverse eukaryotic species (
Figure 3f) underscores its functional importance [
20,
21]. The substitution of a neutral, aromatic tyrosine with a negatively charged aspartic acid (Y975D) likely disrupts critical interactions or local protein structure. This mutation likely perturbs the interaction between SMC-5 and the kleisin subunit NSE-4, which is known to bridge the SMC head domains and recruit NSE-1/3 [
16,
17,
29]. A weakened SMC-5/NSE-4 interaction could destabilize the entire complex specifically on chromatin, leading to the observed partial and dynamic NSE-1 mislocalization. This missense allele thus provides a unique tool to probe the specific role of the SMC-5 C-terminal domain and the Y975 residue in complex stability and NSE-1 association.
Consistent with the essential nature of SMC-5/6, all three novel
smc-5 alleles caused significant defects in fundamental biological processes, including reduced brood size, decreased progeny viability, and slightly increased frequency of male progeny (indicative of X-chromosome nondisjunction) (
Figure 3g–i). These phenotypes confirm that
wsh31,
wsh32, and
wsh33 represent substantial loss-of-function mutations impairing gametogenesis and chromosome segregation fidelity, core functions attributed to SMC-5/6 [
30,
31]. The slightly elevated male frequency seen in
smc-5(wsh31),
smc-5(wsh32), and
smc-5(wsh33) but not in the null
smc-5(ok2421) suggests that X-chromosome nondisjunction is triggered not by a simple loss of SMC-5, but by defective SMC-5 subunits that still enter the SMC-5/6 holocomplex. The nonsense (
wsh31,
wsh32) and missense (
wsh33, Y975D) alleles retain the N-terminal arm yet lack, or disrupt, the C-terminal head that binds the kleisin NSE-4. The severity of the viability defect in the new alleles was comparable to the null mutant, suggesting that even the partial function potentially retained by
smc-5(wsh33) is insufficient for normal embryonic development. It is also important to note that some differences observed in the
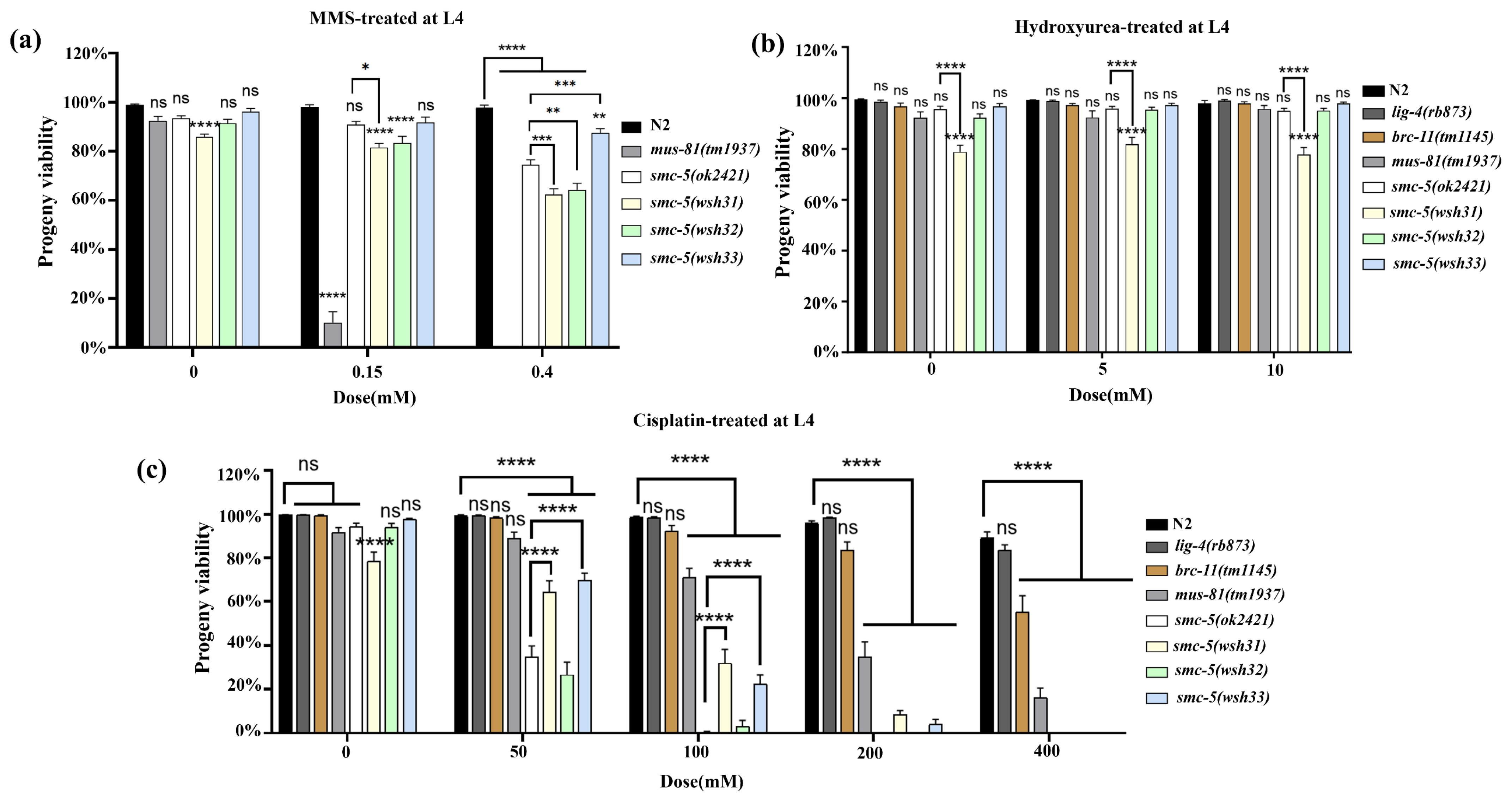
smc-5 mutant phenotypes might arise not solely from the absence of functional SMC-5 protein but also from potential toxicity associated with the different truncated or mutated SMC-5 proteins present in the system. The key differences among the alleles emerged from the DNA damage sensitivity assays. These assays revealed a spectrum of sensitivities, providing insights into the specific roles of SMC-5 domains in coping with different types of DNA lesions. At the L4 stage,
smc-5(wsh33) displayed greater resilience to MMS compared to the truncation alleles
wsh31 and
wsh32, which showed intermediate sensitivity. This suggests that the Y975D mutant protein retains partial function in repairing MMS-induced lesions. However, L1 stage assays, which probe replication-associated repair more directly, revealed profound sensitivity in all
smc-5 mutants, including
wsh33, approaching the null phenotype at higher doses. This highlights the critical role of intact SMC-5/6 during replication for handling alkylation damage. The hypersensitivity of
smc-5 mutants compared to
brc-1(tm1145) (HR deficient) aligns with models where SMC-5/6 acts downstream or in parallel to HR initiation to resolve repair intermediates [
22]. The extreme sensitivity of
mus-81 mutants underscores the importance of structure-specific endonucleases in processing MMS-induced lesions, likely working in concert with SMC-5/6. The insensitivity of
lig-4 mutants confirms the minor role of NHEJ in this context in the germline.
The lack of significant HU sensitivity at the L4 stage is consistent with the fact that pachytene cells, the predominant stage assayed in L4s, are not actively replicating. Conversely, L1 stage exposure revealed moderate HU sensitivity in
smc-5 mutants (
Figure 5b), directly implicating SMC-5/6 in managing stalled replication forks induced by nucleotide depletion [
32,
33]. Interestingly,
smc-5(wsh32) (Q655*) appeared least sensitive among the
smc-5 alleles, suggesting potential specific roles for the N-terminal region versus the C-terminal domain affected in
wsh33. The relative insensitivity of
brc-1 and
lig-4 mutants to HU, contrasted with the sensitivity of
mus-81 and
smc-5 mutants, suggests that resolving stalled/collapsed forks under HU stress primarily requires MUS-81 and SMC-5/6, rather than canonical HR initiation or NHEJ [
23,
24,
25]. Cisplatin induced the most severe phenotypes. All
smc-5 alleles were hypersensitive, as viability at the L1 stage fell to nearly zero even at low doses (
Figure 5c), and the effect was far stronger than in wild-type or in
brc-1 and
lig-4 mutants. This extreme sensitivity underscores that SMC-5/6 is indispensable not only for classical inter-strand cross-link (ICL) repair [
34,
35], but also for resolving DNA–protein cross-links (DPCs), lesions that likewise stall replication. In plants, a recent forward-genetic screen identified SMC6B as a key factor in DPC repair and showed that the SMC-5/6 complex promotes lesion removal through NSE2-dependent SUMOylation at the damage site, acting in parallel to the MUS81 nuclease and WSS1A protease pathways [
36]. The pronounced cisplatin hypersensitivity of our
smc-5 mutants, including the null
smc-5(ok2421) and truncation
smc-5(wsh32), is consistent with this conserved role. This is because cisplatin generates both ICLs and DPC-like adducts that demand coordinated nuclease, protease, and post-translational-modification activities for repair. The slightly milder response of the missense allele
smc-5(wsh33) at the lowest doses suggests residual function, echoing partial-loss alleles in Arabidopsis that retain limited SUMO-ligation capacity. The high sensitivity of
mus-81 mutants in the same assay further supports the idea that SMC-5/6 operates in a pathway that intersects with, but is not redundant to MUS-81-mediated processing of cross-link-associated structures. Analysis of larval development after L1 exposure (
Figure 6) reinforces these points: cisplatin caused a potent, dose-dependent arrest, and
smc-5 mutants were the most compromised. Notably,
smc-5(wsh31) exhibited a persistent L1 arrest even without exogenous damage, implying that SMC-5/6 also mitigates endogenous DPC-like stress during development. Together with the Arabidopsis findings, our data highlight a conserved function for SMC-5/6 in safeguarding replication against both ICLs and DPCs, linking its SUMO-dependent activity directly to developmental progression following genotoxic insult [
34,
35].
Using yeast two-hybrid assays, we previously demonstrated that among the NSEs, only NSE-4 directly interacts with SMC-5. In wild type, NSE-1 and NSE-3 form a tight complex with the kleisin NSE-4, which together attach to the SMC-5/6 ATPase heads [
29]. The Y975D mutation appears to specifically undermine this attachment. Yeast-2-hybrid experiments confirmed that SMC-5(Y975D) no longer binds NSE-4. Thus, Tyr975 of SMC-5 is essential for the SMC5–NSE4 interface that tethers the non-SMC subunits to the core SMC5/6 heterodimer. Our findings provide further validation of another study where yeast two-hybrid and structural analyses in
S. cerevisiae have shown that the Nse4 kleisin bridges the head of Smc5 to the coiled-coil neck of Smc6 [
29,
37]. Structural and cryo-EM studies show that the kleisin subunit NSE4 uses its C-terminal domain to dock onto the bottom “cap” of the SMC5 head, while its N-terminal helix–turn-helix domain binds the head-proximal coiled-coil (“neck”) of SMC6 [
9,
37]. The Y975 residue lies in the region of SMC-5 where Nse4 is known to bind the Smc5 subunit [
37]. This is supported by the 3D modeling of the SMC5–NSE4 interface (
Figure 7f). Replacing this conserved tyrosine with an aspartate likely disrupts a critical contact or local hydrophobic environment needed for the Smc5–Nse4 interaction, thereby uncoupling the kleisin and associated NSE-1/NSE-3 subunits from the ring. Moreover, the interdependency of complex components is further highlighted by recent work showing that loss of
nse-1 leads to mislocalization of NSE-4 in
C. elegans [
10]. Our results present the converse scenario: a defective SMC-5 prevents proper localization of NSE-1 (and by implication NSE-4), underscoring that physical bridging of SMC-5 to NSE-4 is indispensable for holocomplex assembly on chromosomes. Given that mutations in various SMC-5/6 subunits are associated with genome instability disorders and cancer predisposition in humans, understanding the molecular pathology of the SMC-5(Y975D) complex can yield insight into how subtle perturbations in this complex can drive disease phenotypes. For example, our findings imply that therapeutic targeting of the Smc5–Nse4 interaction (or viral proteins that exploit this vulnerability) could severely compromise the complex’s ability to maintain genome integrity. Hence, the convergence of our genetic findings in
C. elegans with human disease data underscores the critical, conserved role of the Smc5–kleisin interface for SMC5/6 complex function.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}