

In Vivo Seeding of Amyloid-β Protein and Implications in Modeling Alzheimer’s Disease Pathology


Abstract
1. Introduction
2. Rodents and Primates as Host Animals for Aβ Seeding
2.1. Genetic Modified Mouse Models
2.2. Rat Models
2.3. Non-Human Primates (NHPs)
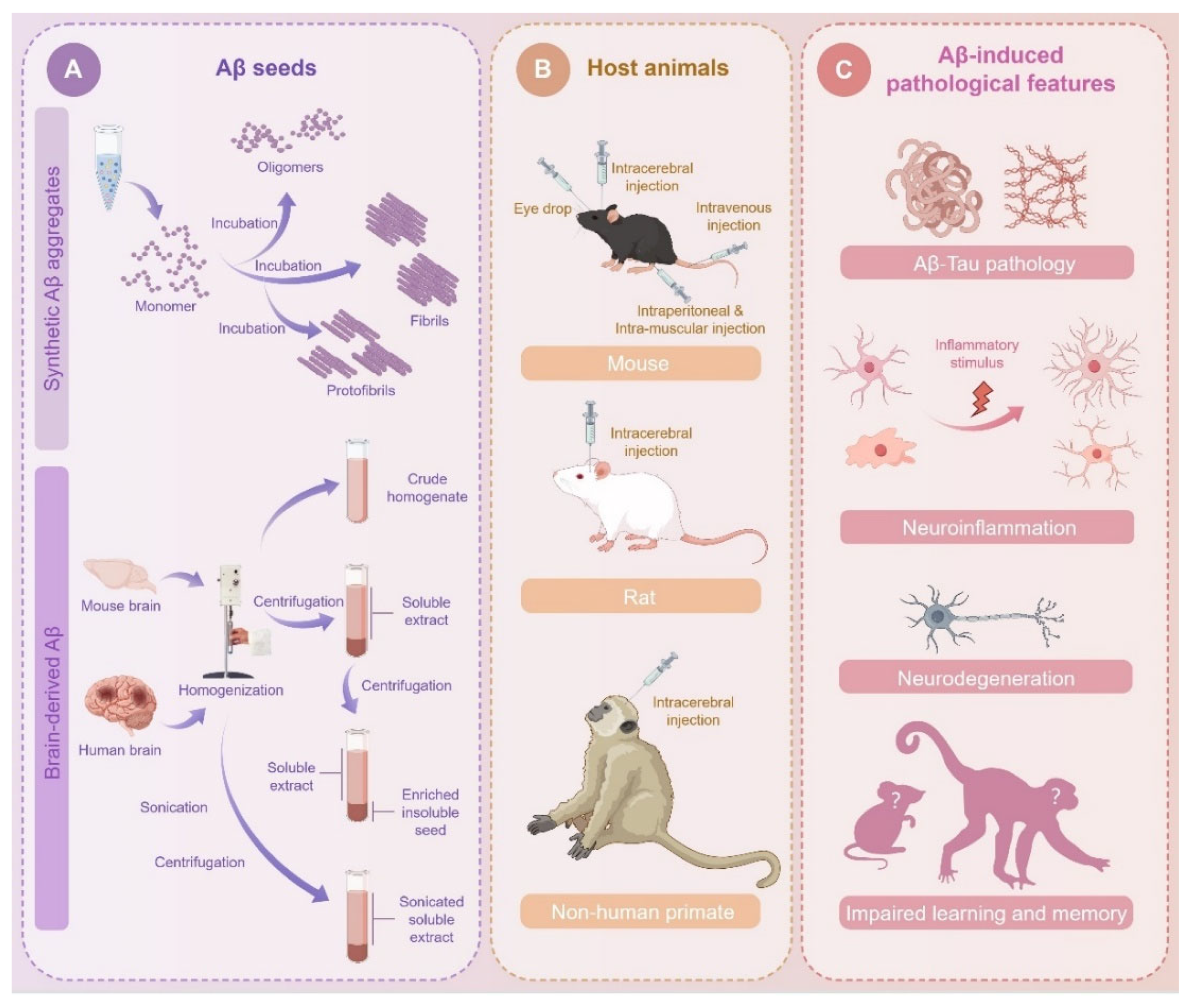
3. Resources of Aβ Seeds
3.1. Synthetic Aβ Aggregates
3.2. Biological Aβ Seeds Derived from Mouse Brain
3.3. Biological Aβ Seeds Derived from Human Brain
3.4. Methodologies for the Preparation of Brain-Derived Aβ Seeds
3.5. Structural Features of Different Types of Aβ Seeds Acquired from in Vitro Synthesis, Mouse Brain, and Human Brain
4. Experimental Strategies for Aβ Inoculation
4.1. Intracerebral Injection
4.2. Peripheral Injection
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eshraghi, M.; Ahmadi, M.; Afshar, S.; Lorzadeh, S.; Adlimoghaddam, A.; Rezvani Jalal, N.; West, R.; Dastghaib, S.; Igder, S.; Torshizi, S.R.N.; et al. Enhancing autophagy in Alzheimer’s disease through drug repositioning. Pharmacol. Ther. 2022, 237, 108171. [Google Scholar] [CrossRef]
- Jia, L.; Du, Y.; Chu, L.; Zhang, Z.; Li, F.; Lyu, D.; Li, Y.; Li, Y.; Zhu, M.; Jiao, H.; et al. Prevalence, risk factors, and management of dementia and mild cognitive impairment in adults aged 60 years or older in China: A cross-sectional study. Lancet Public Health 2020, 5, e661–e671. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Chan, K.Y.; Wang, W.; Wu, J.J.; Liu, L.; Theodoratou, E.; Car, J.; Middleton, L.; Russ, T.C.; Deary, I.J.; Campbell, H.; et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: A systematic review and analysis. Lancet 2013, 381, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Langer, F.; Eisele, Y.S.; Fritschi, S.K.; Staufenbiel, M.; Walker, L.C.; Jucker, M. Soluble Aβ Seeds Are Potent Inducers of Cerebral β-Amyloid Deposition. J. Neurosci. 2011, 31, 14488–14495. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Rijal Upadhaya, A.; Kosterin, I.; Kumar, S.; Von Arnim, C.A.F.; Yamaguchi, H.; Fändrich, M.; Walter, J.; Thal, D.R. Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 2014, 137, 887–903. [Google Scholar] [CrossRef]
- Carreiras, M.; Mendes, E.; Perry, M.; Francisco, A.; Marco-Contelles, J. The Multifactorial Nature of Alzheimer’s Disease for Developing Potential Therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Age-Associated Changes in the Immune System and Blood–Brain Barrier Functions. Int. J. Mol. Sci. 2019, 20, 1632. [Google Scholar] [CrossRef]
- Lukiw, W.J. MicroRNA (miRNA) Complexity in Alzheimer’s Disease (AD). Biology 2023, 12, 788. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Banik, A.; Brown, R.E.; Bamburg, J.; Lahiri, D.K.; Khurana, D.; Friedland, R.P.; Chen, W.; Ding, Y.; Mudher, A.; Padjen, A.L.; et al. Translation of Pre-Clinical Studies into Successful Clinical Trials for Alzheimer’s Disease: What are the Roadblocks and How Can They Be Overcome? J. Alzheimer’s Dis. 2015, 47, 815–843. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Pang, K.; Jiang, R.; Zhang, W.; Yang, Z.; Li, L.L.; Shimozawa, M.; Tambaro, S.; Mayer, J.; Zhang, B.; Li, M.; et al. An App knock-in rat model for Alzheimer’s disease exhibiting Aβ and tau pathologies, neuronal death and cognitive impairments. Cell Res. 2022, 32, 157–175. [Google Scholar] [CrossRef]
- He, Z.; Zhang, W.; Chen, P.; Li, S.; Tao, M.; Yue, F.; Hong, W.; Feng, S.; Jing, N. Amyloid-β oligomers drive amyloid deposit and cascaded tau pathology of Alzheimer’s disease in aged brains of non-human primates. J. Genet. Genom. 2025. [Google Scholar] [CrossRef]
- Baerends, E.; Soud, K.; Folke, J.; Pedersen, A.K.; Henmar, S.; Konrad, L.; Lycas, M.D.; Mori, Y.; Pakkenberg, B.; Woldbye, D.P.D.; et al. Modeling the early stages of Alzheimer’s disease by administering intracerebroventricular injections of human native Aβ oligomers to rats. Acta Neuropathol. Commun. 2022, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Bravo-Alegria, J.; Moreno-Gonzalez, I.; Duran-Aniotz, C.; Gamez, N.; Edwards Iii, G.; Soto, C. Transmission of cerebral amyloid pathology by peripheral administration of misfolded Aβ aggregates. Mol. Psychiatry 2021, 26, 5690–5701. [Google Scholar] [CrossRef]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Trujillo-Estrada, L.; Sanchez-Mejias, E.; Sanchez-Varo, R.; Garcia-Leon, J.A.; Nuñez-Diaz, C.; Davila, J.C.; Vitorica, J.; Laferla, F.M.; Moreno-Gonzalez, I.; Gutierrez, A.; et al. Animal and Cellular Models of Alzheimer’s Disease: Progress, Promise, and Future Approaches. Neurosci. 2022, 28, 572–593. [Google Scholar] [CrossRef] [PubMed]
- Herard, A.S.; Petit, F.; Gary, C.; Guillermier, M.; Boluda, S.; Garin, C.M.; The Brainbank Neuro-CEB Neuropathology Network; Lam, S.; Dhenain, M. Induction of amyloid-β deposits from serially transmitted, histologically silent, Aβ seeds issued from human brains. Acta Neuropathol. Commun. 2020, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Kechko, O.I.; Adzhubei, A.A.; Makarov, A.A.; Mitkevich, V.A. Switching On/Off Amyloid Plaque Formation in Transgenic Animal Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 25, 72. [Google Scholar] [CrossRef]
- Xu, G.; Ran, Y.; Fromholt, S.E.; Fu, C.; Yachnis, A.T.; Golde, T.E.; Borchelt, D.R. Murine Aβ over-production produces diffuse and compact Alzheimer-type amyloid deposits. Acta Neuropathol. Commun. 2015, 3, 72. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Younkin, L.H.; Gonzales, V.; Fadale, D.J.; Slunt, H.H.; Lester, H.A.; Younkin, S.G.; Borchelt, D.R. Rodent Aβ Modulates the Solubility and Distribution of Amyloid Deposits in Transgenic Mice. J. Biol. Chem. 2007, 282, 22707–22720. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- Qian, Z.; Li, Y.; Ye, K. Advancements and challenges in mouse models of Alzheimer’s disease. Trends Mol. Med. 2024, 30, 1152–1164. [Google Scholar] [CrossRef]
- Zhong, M.Z.; Peng, T.; Duarte, M.L.; Wang, M.; Cai, D. Updates on mouse models of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 23. [Google Scholar] [CrossRef]
- Dodart, J.-C.; Meziane, H.; Mathis, C.; Bales, K.R.; Paul, S.M.; Ungerer, A. Behavioral disturbances in transgenic mice overexpressing the V717F Β-amyloid precursor protein. Behav. Neurosci. 1999, 113, 982–990. [Google Scholar] [CrossRef]
- Hartman, R.E.; Izumi, Y.; Bales, K.R.; Paul, S.M.; Wozniak, D.F.; Holtzman, D.M. Treatment with an Amyloid-β Antibody Ameliorates Plaque Load, Learning Deficits, and Hippocampal Long-Term Potentiation in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2005, 25, 6213–6220. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, A.; Vickers, J.C.; Adlard, P.A.; Dickson, T.C. Dystrophic neurites in TgCRND8 and Tg2576 mice mimic human pathological brain aging. Neurobiol. Aging 2009, 30, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.H.; Bondolfi, L.; Hunziker, D.; Schlecht, H.P.; Carver, K.; Maguire, E.; Abramowski, D.; Wiederhold, K.H.; Sturchler-Pierrat, C.; Jucker, M.; et al. Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol. Aging 2003, 24, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.-S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset Amyloid Deposition and Cognitive Deficits in Transgenic Mice Expressing a Double Mutant Form of Amyloid Precursor Protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Dudal, S.; Krzywkowski, P.; Paquette, J.; Morissette, C.; Lacombe, D.; Tremblay, P.; Gervais, F. Inflammation occurs early during the Aβ deposition process in TgCRND8 mice. Neurobiol. Aging 2004, 25, 861–871. [Google Scholar] [CrossRef]
- Janus, C.; Flores, A.Y.; Xu, G.; Borchelt, D.R. Behavioral abnormalities in APPSwe/PS1dE9 mouse model of AD-like pathology: Comparative analysis across multiple behavioral domains. Neurobiol. Aging 2015, 36, 2519–2532. [Google Scholar] [CrossRef]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in Amyloid-β and Tau in the Cerebrospinal Fluid of Transgenic Mice Overexpressing Amyloid Precursor Protein. Sci. Transl. Med. 2013, 5, 194re2. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Kang, Z.; Pei, G.; Le, Y. Amyloid Deposition and Inflammation in APPswe/PS1dE9 Mouse Model of Alzheimers Disease. Curr. Alzheimer Res. 2009, 6, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Richard, B.C.; Kurdakova, A.; Baches, S.; Bayer, T.A.; Weggen, S.; Wirths, O. Gene Dosage Dependent Aggravation of the Neurological Phenotype in the 5XFAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; Laferla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Masuda, A.; Kobayashi, Y.; Kogo, N.; Saito, T.; Saido, T.C.; Itohara, S. Cognitive deficits in single App knock-in mouse models. Neurobiol. Learn. Mem. 2016, 135, 73–82. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-β-associated cognitive impairment. J. Alzheimers Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric Aβ, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Le Merre, P.; Ahrlund-Richter, S.; Carlen, M. The mouse prefrontal cortex: Unity in diversity. Neuron 2021, 109, 1925–1944. [Google Scholar] [CrossRef]
- Jobson, D.D.; Hase, Y.; Clarkson, A.N.; Kalaria, R.N. The role of the medial prefrontal cortex in cognition, ageing and dementia. Brain Commun. 2021, 3, fcab125. [Google Scholar] [CrossRef]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 2021, 15, 652111. [Google Scholar] [CrossRef]
- Ennerfelt, H.E.; Lukens, J.R. The role of innate immunity in Alzheimer’s disease. Immunol. Rev. 2020, 297, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Farberg, A.S.; Gearing, M.; Dooyema, J.; Long, P.M.; Anderson, D.C.; Davis-Turak, J.; Coppola, G.; Geschwind, D.H.; Pare, J.F.; et al. Tauopathy with paired helical filaments in an aged chimpanzee. J. Comp. Neurol. 2008, 509, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Gearing, M.; Tigges, J.; Mori, H.; Mirra, S.S. β-Amyloid (Aβ) Deposition in the Brains of Aged Orangutans. Neurobiol. Aging 1997, 18, 139–146. [Google Scholar] [CrossRef]
- Edler, M.K.; Sherwood, C.C.; Meindl, R.S.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Mufson, E.J.; Hof, P.R.; Raghanti, M.A. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol. Aging 2017, 59, 107–120. [Google Scholar] [CrossRef]
- Heuer, E.; Rosen, R.F.; Cintron, A.; Walker, L.C. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr. Pharm. Des. 2012, 18, 1159–1169. [Google Scholar] [CrossRef]
- Edler, M.K.; Munger, E.L.; Meindl, R.S.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Mufson, E.J.; Hof, P.R.; Sherwood, C.C.; Raghanti, M.A. Neuron loss associated with age but not Alzheimer’s disease pathology in the chimpanzee brain. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190619. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-H.; He, X.-P.; Li, H.; He, R.-Q.; Hu, X.-T. Age-associated changes in amyloid-β and formaldehyde concentrations in cerebrospinal fluid of rhesus monkeys. Zool. Res. 2020, 41, 444–448. [Google Scholar] [CrossRef]
- Stonebarger, G.; Urbanski, H.; Woltjer, R.; Vaughan, K.; Ingram, D.; Schultz, P.; Calderazzo, S.; Siedeman, J.; Mattison, J.; Rosene, D.; et al. Amyloidosis increase is not attenuated by long-term calorie restriction or related to neuron density in the prefrontal cortex of extremely aged rhesus macaques. GeroScience 2020, 42, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, B.; Lu, J.; Wu, Y.; Wang, S.; Yao, Z.; Zhu, L.; Qiao, Y.; Sun, Q.; Qin, W.; et al. Brains of rhesus monkeys display Aβ deposits and glial pathology while lacking Aβ dimers and other Alzheimer’s pathologies. Aging Cell 2019, 18, e12978. [Google Scholar] [CrossRef]
- Rhesus Macaque Genome Sequencing and Analysis Consortium; Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Wibom, M.; Pawlik, D.; Englund, E.; Hansson, O. Correlation of In Vivo [18F]Flortaucipir With Postmortem Alzheimer Disease Tau Pathology. JAMA Neurol. 2019, 76, 310–317. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Leslie, S.; Yang, S.T.; Wang, M.; Nairn, A.C. Alzheimer’s-like pathology in aging rhesus macaques: Unique opportunity to study the etiology and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 26230–26238. [Google Scholar] [CrossRef]
- Moore, T.L.; Killiany, R.J.; Herndon, J.G.; Rosene, D.L.; Moss, M.B. Executive system dysfunction occurs as early as middle-age in the rhesus monkey. Neurobiol. Aging 2006, 27, 1484–1493. [Google Scholar] [CrossRef]
- Moore, T.L.; Killiany, R.J.; Herndon, J.G.; Rosene, D.L.; Moss, M.B. Impairment in abstraction and set shifting in aged rhesus monkeys. Neurobiol. Aging 2003, 24, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Herndon, J.G.; Moss, M.B.; Rosene, D.L.; Killiany, R.J. Patterns of cognitive decline in aged rhesus monkeys. Behav. Brain Res. 1997, 87, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Shively, C.A.; Keene, C.D.; Jorgensen, M.J.; Andrews, R.N.; Register, T.C.; Montine, T.J.; Wilson, A.M.; Neth, B.J.; Mintz, A.; et al. A nonhuman primate model of early Alzheimer’s disease pathologic change: Implications for disease pathogenesis. Alzheimer’s Dement. 2019, 15, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Ott, S.; Donis-Cox, K.; Janssen, W.G.; Bliss-Moreau, E.; Rudebeck, P.H.; Baxter, M.G.; Morrison, J.H. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc. Natl. Acad. Sci. USA 2019, 116, 26239–26246. [Google Scholar] [CrossRef]
- Beckman, D.; Chakrabarty, P.; Ott, S.; Dao, A.; Zhou, E.; Janssen, W.G.; Donis-Cox, K.; Muller, S.; Kordower, J.H.; Morrison, J.H. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimer’s Dement. 2021, 17, 933–945. [Google Scholar] [CrossRef]
- Tu, Z.; Yan, S.; Han, B.; Li, C.; Liang, W.; Lin, Y.; Ding, Y.; Wei, H.; Wang, L.; Xu, H.; et al. Tauopathy promotes spinal cord-dependent production of toxic amyloid-beta in transgenic monkeys. Signal Transduct. Target. Ther. 2023, 8, 358. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous Induction of Cerebral ß-Amyloidogenesis Is Governed by Agent and Host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Ulm, B.S.; Borchelt, D.R.; Moore, B.D. Remodeling Alzheimer-amyloidosis models by seeding. Mol. Neurodegener. 2021, 16, 8. [Google Scholar] [CrossRef]
- Stohr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef]
- Li, W.; Wu, Y.; Min, F.; Li, Z.; Huang, J.; Huang, R. A nonhuman primate model of Alzheimer’s disease generated by intracranial injection of amyloid-beta42 and thiorphan. Metab. Brain Dis. 2010, 25, 277–284. [Google Scholar] [CrossRef]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Induction of β(A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol. Neurobiol. 1994, 8, 25–39. [Google Scholar] [CrossRef]
- Parvin, F.; Larsson, J.N.K.; Jackson, W.S.; Nyström, S.; Hammarström, P. Efficient Seeding of Cerebral Vascular Aβ-Amyloidosis by Recombinant AβM1-42 Amyloid Fibrils. J. Mol. Biol. 2025, 437, 168923. [Google Scholar] [CrossRef]
- Nagao, M.; Hatae, A.; Mine, K.; Tsutsumi, S.; Omori, H.; Hirata, M.; Arimatsu, M.; Taniguchi, C.; Watanabe, T.; Kubota, K.; et al. The Effects of Ninjinyoeito on Impaired Spatial Memory and Prefrontal Cortical Synaptic Plasticity through α-Amino-3-hydroxy-5-4-isoxazole Propionic Acid Receptor Subunit in a Rat Model with Cerebral Ischemia and β-Amyloid Injection. Evid.-Based Complement. Altern. Med. 2023, 2023, 6035589. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Yamano, S.; Imagawa, N.; Igami, K.; Miyazaki, T.; Ito, H.; Watanabe, T.; Kubota, K.; Katsurabayashi, S.; Iwasaki, K. Effect of Lactobacillus paracasei A221-fermented ginseng on impaired spatial memory in a rat model with cerebral ischemia and β-amyloid injection. Tradit. Kampo Med. 2019, 6, 96–104. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Silva, N.M.L.E.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.B.; et al. Alzheimer’s Disease-Like Pathology Induced by Amyloid-β Oligomers in Nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Feng, S.; Lu, C.; Zhang, T.; Tao, G.; Liu, J.; Yue, C.; Jing, N. Synthetic amyloid-β oligomers drive early pathological progression of Alzheimer’s disease in nonhuman primates. iScience 2021, 24, 103207. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of β-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.; Portelius, E.; Zetterberg, H.; et al. Identification of neurotoxic cross-linked amyloid-β dimers in Alzheimer brain. Brain 2019, 142, 1441–1457. [Google Scholar] [CrossRef]
- Mc Donald, J.M.; O’Malley, T.T.; Liu, W.; Mably, A.J.; Brinkmalm, G.; Portelius, E.; Wittbold, W.M., 3rd; Frosch, M.P.; Walsh, D.M. The aqueous phase of Alzheimer’s disease brain contains assemblies built from ~4 and ~7 kDa Aβ species. Alzheimers Dement. 2015, 11, 1286–1305. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Stohr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stohr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Cukalevski, R.; Yang, X.; Meisl, G.; Weininger, U.; Bernfur, K.; Frohm, B.; Knowles, T.P.J.; Linse, S. The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 2015, 6, 4215–4233. [Google Scholar] [CrossRef]
- Pauwels, K.; Williams, T.L.; Morris, K.L.; Jonckheere, W.; Vandersteen, A.; Kelly, G.; Schymkowitz, J.; Rousseau, F.; Pastore, A.; Serpell, L.C.; et al. Structural basis for increased toxicity of pathological Aβ42:Aβ40 ratios in Alzheimer disease. J. Biol. Chem. 2012, 287, 5650–5660. [Google Scholar] [CrossRef]
- Meng, F.; Kim, J.Y.; Louis, J.M.; Chung, H.S. Single-Molecule Characterization of Heterogeneous Oligomer Formation during Co-Aggregation of 40- and 42-Residue Amyloid-β. J. Am. Chem. Soc. 2024, 146, 24426–24439. [Google Scholar] [CrossRef]
- Chang, H.W.; Ma, H.I.; Wu, Y.S.; Lee, M.C.; Chung-Yueh Yuan, E.; Huang, S.J.; Cheng, Y.S.; Wu, M.H.; Tu, L.H.; Chan, J.C.C. Site specific NMR characterization of abeta-40 oligomers cross seeded by abeta-42 oligomers. Chem. Sci. 2022, 13, 8526–8535. [Google Scholar] [CrossRef]
- Jan, A.; Gokce, O.; Luthi-Carter, R.; Lashuel, H.A. The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 2008, 283, 28176–28189. [Google Scholar] [CrossRef]
- Hasegawa, K.; Yamaguchi, I.; Omata, S.; Gejyo, F.; Naiki, H. Interaction between Aβ(1-42) and Aβ(1-40) in Alzheimer’s β-amyloid fibril formation in vitro. Biochemistry 1999, 38, 15514–15521. [Google Scholar] [CrossRef]
- Wang, L.; Park, S.; Choi, J.H.; Lee, C.Y.; Eom, K.; Kwon, T. Molecular insight into cross-interaction between amyloid β isoforms and its effect on aggregation pathways. J. Biomol. Struct. Dyn. 2025. [Google Scholar] [CrossRef]
- Shen, Y.X.; Wei, W.; Xu, S.Y. Protective effects of melatonin on cortico-hippocampal neurotoxicity induced by amyloid beta-peptide 25-35. Acta Pharmacol. Sin. 2002, 23, 71–76. [Google Scholar]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid β-peptide 25–35 self-assembly and its inhibition: A model undecapeptide system to gain atomistic and secondary structure details of the Alzheimer’s disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, L.; Zheng, Q.; Song, Y.; Huang, W.; Yang, L.; Xiong, Y.; Cai, Z.; Chen, Y.; Yuan, J. Kai-xin-san improves cognitive impairment in D-gal and Aβ25-35 induced ad rats by regulating gut microbiota and reducing neuronal damage. J. Ethnopharmacol. 2024, 329, 118161. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Konishi, M.; Higashi, Y.; Saito, M.; Akizawa, T. Five-mer peptides prevent short-term spatial memory deficits in Aβ25-35-induced Alzheimer’s model mouse by suppressing Aβ25-35 aggregation and resolving its aggregate form. Alzheimer’s Res. Ther. 2023, 15, 83. [Google Scholar] [CrossRef]
- Stine, W.B., Jr.; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y.; Feng, Y.; Liu, H.; Shen, X.; Chen, K.; Ma, J.; Jiang, H. Inhibitor discovery targeting the intermediate structure of β-amyloid peptide on the conformational transition pathway: Implications in the aggregation mechanism of β-amyloid peptide. Biochemistry 2006, 45, 10963–10972. [Google Scholar] [CrossRef]
- Schrempel, S.; Kottwitz, A.K.; Piechotta, A.; Gnoth, K.; Büschgens, L.; Hartlage-Rübsamen, M.; Morawski, M.; Schenk, M.; Kleinschmidt, M.; Serrano, G.E.; et al. Identification of isoAsp7-Aβ as a major Aβ variant in Alzheimer’s disease, dementia with Lewy bodies and vascular dementia. Acta Neuropathol. 2024, 148, 78. [Google Scholar] [CrossRef]
- Kozin, S.A.; Mitkevich, V.A.; Makarov, A.A. Amyloid-β containing isoaspartate 7 as potential biomarker and drug target in Alzheimer’s disease. Mendeleev Commun. 2016, 26, 269–275. [Google Scholar] [CrossRef]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally Applied Synthetic Peptide isoAsp7-Aβ(1-42) Triggers Cerebral β-Amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Åslund, A.; Hammarström, P.; Nilsson, K.P.R.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Bolmont, T.; Heikenwalder, M.; Langer, F.; Jacobson, L.H.; Yan, Z.X.; Roth, K.; Aguzzi, A.; Staufenbiel, M.; Walker, L.C.; et al. Induction of cerebral β-amyloidosis: Intracerebral versus systemic Aβ inoculation. Proc. Natl. Acad. Sci. USA 2009, 106, 12926–12931. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Fritschi, S.K.; Hamaguchi, T.; Obermuller, U.; Fuger, P.; Skodras, A.; Schafer, C.; Odenthal, J.; Heikenwalder, M.; Staufenbiel, M.; et al. Multiple Factors Contribute to the Peripheral Induction of Cerebral β-Amyloidosis. J. Neurosci. 2014, 34, 10264–10273. [Google Scholar] [CrossRef] [PubMed]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. 2020, 30, 36–45. [Google Scholar] [CrossRef]
- Roos, T.T.; Garcia, M.G.; Martinsson, I.; Mabrouk, R.; Israelsson, B.; Deierborg, T.; Kobro-Flatmoen, A.; Tanila, H.; Gouras, G.K. Neuronal spreading and plaque induction of intracellular Aβ and its disruption of Aβ homeostasis. Acta Neuropathol. 2021, 142, 669–687. [Google Scholar] [CrossRef]
- Aires, V.; Ziegler-Waldkirch, S.; Friesen, M.; Reichardt, W.; Erny, D.; Loreth, D.; Harborne, A.; Kretz, O.; Von Elverfeldt, D.; Meyer-Luehmann, M. Seed-induced Aβ deposits in the corpus callosum disrupt white matter integrity in a mouse model of Alzheimer’s disease. Front. Cell. Neurosci. 2022, 16, 862918. [Google Scholar] [CrossRef]
- Fritschi, S.K.; Cintron, A.; Ye, L.; Mahler, J.; Buhler, A.; Baumann, F.; Neumann, M.; Nilsson, K.P.; Hammarstrom, P.; Walker, L.C.; et al. Aβ seeds resist inactivation by formaldehyde. Acta Neuropathol. 2014, 128, 477–484. [Google Scholar] [CrossRef]
- Ye, L.; Rasmussen, J.; Kaeser, S.A.; Marzesco, A.M.; Obermuller, U.; Mahler, J.; Schelle, J.; Odenthal, J.; Kruger, C.; Fritschi, S.K.; et al. Aβ seeding potency peaks in the early stages of cerebral β-amyloidosis. EMBO Rep. 2017, 18, 1536–1544. [Google Scholar] [CrossRef]
- Ruiz-Riquelme, A.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Burwinkel, M.; Lutzenberger, M.; Heppner, F.L.; Schulz-Schaeffer, W.; Baier, M. Intravenous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol. Commun. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Fritschi, S.K.; Schelle, J.; Obermuller, U.; Degenhardt, K.; Kaeser, S.A.; Eisele, Y.S.; Walker, L.C.; Baumann, F.; Staufenbiel, M.; et al. Persistence of Aβ seeds in APP null mouse brain. Nat. Neurosci. 2015, 18, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Eisele, Y.S.; Varvel, N.H.; Lamb, B.T.; Walker, L.C.; Jucker, M. The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in the brain. Acta Neuropathol. 2012, 123, 31–37. [Google Scholar] [CrossRef]
- Walker, L.C.; Callahan, M.J.; Bian, F.; Durham, R.A.; Roher, A.E.; Lipinski, W.J. Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides 2002, 23, 1241–1247. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Grillo, S.K.; Lemus, A.; DeArmond, S.J.; Prusiner, S.B. Bioluminescence imaging of Aβ deposition in bigenic mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2011, 108, 2528–2533. [Google Scholar] [CrossRef]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Neurodegeneration: Amyloid-β pathology induced in humans. Nature 2015, 525, 193–194. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Seeds of Dementia. Sci. Am. 2013, 308, 52–57. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.; Thal, D.R.; Van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-Dependent Changes in Brain, CSF, and Plasma Amyloid β Protein in the Tg2576 Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2001, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kokjohn, T.A.; Roher, A.E. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: Understanding the paradigms, limitations, and contributions. Alzheimer’s Dement. 2009, 5, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-W.; Tanzi, R.E. Presenilins and Alzheimer’s disease. Curr. Opin. Neurobiol. 1997, 7, 683–688. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef]
- Andersson, E.; Schultz, N.; Saito, T.; Saido, T.C.; Blennow, K.; Gouras, G.K.; Zetterberg, H.; Hansson, O. Cerebral Aβ deposition precedes reduced cerebrospinal fluid and serum Aβ42/Aβ40 ratios in the AppNL−F/NL−F knock-in mouse model of Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 64. [Google Scholar] [CrossRef]
- Garai, K.; Verghese, P.B.; Baban, B.; Holtzman, D.M.; Frieden, C. The binding of apolipoprotein E to oligomers and fibrils of amyloid-β alters the kinetics of amyloid aggregation. Biochemistry 2014, 53, 6323–6331. [Google Scholar] [CrossRef]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e1023. [Google Scholar] [CrossRef]
- Milani, R.; Mazzeo, L.A.; Vismara, D.; Salemi, I.; Dainese, E.; Maderna, E.; Pellencin, E.; Catania, M.; Campanella, N.; Di Fede, G.; et al. Spontaneous intracerebral haemorrhage associated with early-onset cerebral amyloid angiopathy and Alzheimer’s disease neuropathological changes five decades after cadaveric dura mater graft. Acta Neuropathol. Commun. 2023, 11, 30. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Barria, M.A.; Peden, A.H.; Yull, H.M.; Kirkpatrick, J.; Adlard, P.; Ironside, J.W.; Head, M.W. UK Iatrogenic Creutzfeldt–Jakob disease: Investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches. Acta Neuropathol. 2017, 133, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Farmer, S.F.; Hyare, H.; Jaunmuktane, Z.; Mead, S.; Ryan, N.S.; Schott, J.M.; Werring, D.J.; Rudge, P.; Collinge, J. Iatrogenic Alzheimer’s disease in recipients of cadaveric pituitary-derived growth hormone. Nat. Med. 2024, 30, 394–402. [Google Scholar] [CrossRef]
- Li, X.; Ospitalieri, S.; Robberechts, T.; Hofmann, L.; Schmid, C.; Rijal Upadhaya, A.; Koper, M.J.; Von Arnim, C.A.F.; Kumar, S.; Willem, M.; et al. Seeding, maturation and propagation of amyloid β-peptide aggregates in Alzheimer’s disease. Brain 2022, 145, 3558–3570. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Mahler, J.; Beschorner, N.; Kaeser, S.A.; Häsler, L.M.; Baumann, F.; Nyström, S.; Portelius, E.; Blennow, K.; Lashley, T.; et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 13018–13023. [Google Scholar] [CrossRef]
- Walker, L.C.; Levine, H. Corruption and Spread of Pathogenic Proteins in Neurodegenerative Diseases. J. Biol. Chem. 2012, 287, 33109–33115. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef]
- Lam, S.; Hérard, A.-S.; Boluda, S.; Petit, F.; Eddarkaoui, S.; Cambon, K.; Letournel, F.; Martin-Négrier, M.-L.; Faisant, M.; Godfraind, C.; et al. Pathological changes induced by Alzheimer’s brain inoculation in amyloid-beta plaque-bearing mice. Acta Neuropathol. Commun. 2022, 10, 112. [Google Scholar] [CrossRef]
- Lam, S.; Petit, F.; Herard, A.S.; Boluda, S.; Eddarkaoui, S.; Guillermier, M.; The Brain Bank Neuro–Cognitive and Experimental Biology Neuropathology Network; Buee, L.; Duyckaerts, C.; Haik, S.; et al. Transmission of amyloid-beta and tau pathologies is associated with cognitive impairments in a primate. Acta Neuropathol. Commun. 2021, 9, 165. [Google Scholar] [CrossRef]
- Gary, C.; Lam, S.; Hérard, A.-S.; Koch, J.E.; Petit, F.; Gipchtein, P.; Sawiak, S.J.; Caillierez, R.; Eddarkaoui, S.; Colin, M.; et al. Encephalopathy induced by Alzheimer brain inoculation in a non-human primate. Acta Neuropathol. Commun. 2019, 7, 126. [Google Scholar] [CrossRef]
- Fritschi, S.K.; Langer, F.; Kaeser, S.A.; Maia, L.F.; Portelius, E.; Pinotsi, D.; Kaminski, C.F.; Winkler, D.T.; Maetzler, W.; Keyvani, K.; et al. Highly potent soluble amyloid-β seeds in human Alzheimer brain but not cerebrospinal fluid. Brain 2014, 137, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Jin, M.; Willem, M.; Haass, C.; Frosch, M.P.; Walsh, D.M. Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol. 2018, 136, 19–40. [Google Scholar] [CrossRef]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Esparza, T.J.; Leduc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Yagi-Utsumi, M.; Kato, K. Conformational Variability of Amyloid-β and the Morphological Diversity of Its Aggregates. Molecules 2022, 27, 4787. [Google Scholar] [CrossRef]
- Niu, Z.; Gui, X.; Feng, S.; Reif, B. Aggregation Mechanisms and Molecular Structures of Amyloid-β in Alzheimer’s Disease. Chem. A Eur. J. 2024, 30, e202400277. [Google Scholar] [CrossRef]
- Askenazi, M.; Kavanagh, T.; Pires, G.; Ueberheide, B.; Wisniewski, T.; Drummond, E. Compilation of reported protein changes in the brain in Alzheimer’s disease. Nat. Commun. 2023, 14, 4466. [Google Scholar] [CrossRef]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; Desousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020, 139, 503–526. [Google Scholar] [CrossRef]
- Morales, R.; Duran-Aniotz, C.; Castilla, J.; Estrada, L.D.; Soto, C. De novo induction of amyloid-β deposition in vivo. Mol. Psychiatry 2012, 17, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Fritz, J.J.; Dooyema, J.; Cintron, A.F.; Hamaguchi, T.; Lah, J.J.; LeVine, H., 3rd; Jucker, M.; Walker, L.C. Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J. Neurochem. 2012, 120, 660–666. [Google Scholar] [CrossRef]
- Baker, H.F.; Ridley, R.M.; Duchen, L.W.; Crow, T.J.; Bruton, C.J. Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int. J. Exp. Pathol. 1993, 74, 441–454. [Google Scholar] [PubMed]
- Maclean, C.J.; Baker, H.F.; Ridley, R.M.; Mori, H. Naturally occurring and experimentally induced β-amyloid deposits in the brains of marmosets (Callithrix jacchus). J. Neural Transm. 2000, 107, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef]
- Mandal, P.K.; Pettegrew, J.W.; Masliah, E.; Hamilton, R.L.; Mandal, R. Interaction between Aβ peptide and α synuclein: Molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem. Res. 2006, 31, 1153–1162. [Google Scholar] [CrossRef]
- Chia, S.; Flagmeier, P.; Habchi, J.; Lattanzi, V.; Linse, S.; Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 8005–8010. [Google Scholar] [CrossRef]
- Iljina, M.; Dear, A.J.; Garcia, G.A.; De, S.; Tosatto, L.; Flagmeier, P.; Whiten, D.R.; Michaels, T.C.T.; Frenkel, D.; Dobson, C.M.; et al. Quantifying Co-Oligomer Formation by α-Synuclein. ACS Nano 2018, 12, 10855–10866. [Google Scholar] [CrossRef]
- Ziegler-Waldkirch, S.; D′Errico, P.; Sauer, J.F.; Erny, D.; Savanthrapadian, S.; Loreth, D.; Katzmarski, N.; Blank, T.; Bartos, M.; Prinz, M.; et al. Seed-induced Aβ deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer’s disease. EMBO J. 2018, 37, 167–182. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fa, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2019, 68, 415. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, Q.; Xiao, L.; Huang, Y.; Yang, Z.; Wu, Z.; Tu, Z.; Qin, W.; Chen, H.; Wu, D.; et al. Effects of ultrasound on functional properties, structure and glycation properties of proteins: A review. Crit. Rev. Food Sci. Nutr. 2021, 61, 2471–2481. [Google Scholar] [CrossRef]
- Hong, W.; Liu, W.; Desousa, A.O.; Young-Pearse, T.; Walsh, D.M. Methods for the isolation and analysis of Aβ from postmortem brain. Front. Neurosci. 2023, 17, 1108715. [Google Scholar] [CrossRef] [PubMed]
- Esparza, T.J.; Wildburger, N.C.; Jiang, H.; Gangolli, M.; Cairns, N.J.; Bateman, R.J.; Brody, D.L. Soluble Amyloid-beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep. 2016, 6, 38187. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Walti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Bockmann, A.; Guntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Xiao, Y.; Matsuda, I.; Inoue, M.; Sasahara, T.; Hoshi, M.; Ishii, Y. NMR-based site-resolved profiling of β-amyloid misfolding reveals structural transitions from pathologically relevant spherical oligomer to fibril. J. Biol. Chem. 2020, 295, 458–467. [Google Scholar] [CrossRef]
- Yu, L.; Edalji, R.; Harlan, J.E.; Holzman, T.F.; Lopez, A.P.; Labkovsky, B.; Hillen, H.; Barghorn, S.; Ebert, U.; Richardson, P.L.; et al. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry 2009, 48, 1870–1877. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Qahwash, I.; Leapman, R.D.; Meredith, S.C.; Tycko, R. Seeded growth of β-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. USA 2009, 106, 7443–7448. [Google Scholar] [CrossRef]
- Ghosh, U.; Thurber, K.R.; Yau, W.M.; Tycko, R. Molecular structure of a prevalent amyloid-β fibril polymorph from Alzheimer’s disease brain tissue. Proc. Natl. Acad. Sci. USA 2021, 118, e2023089118. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fandrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lovestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, W.; Murzin, A.G.; Schweighauser, M.; Huang, M.; Lovestam, S.; Peak-Chew, S.Y.; Saito, T.; Saido, T.C.; Macdonald, J.; et al. Cryo-EM structures of amyloid-β filaments with the Arctic mutation (E22G) from human and mouse brains. Acta Neuropathol. 2023, 145, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Murzin, A.G.; Peak-Chew, S.; Franco, C.; Garringer, H.J.; Newell, K.L.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of Aβ40 filaments from the leptomeninges of individuals with Alzheimer’s disease and cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2023, 11, 191. [Google Scholar] [CrossRef]
- Fernandez, A.; Hoq, M.R.; Hallinan, G.I.; Li, D.; Bharath, S.R.; Vago, F.S.; Zhang, X.; Ozcan, K.A.; Newell, K.L.; Garringer, H.J.; et al. Cryo-EM structures of amyloid-β and tau filaments in Down syndrome. Nat. Struct. Mol. Biol. 2024, 31, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.C.; Roy, J.; Chau, S.C.; Wong, K.H.; Shi, L.; Poon, C.H.; Wang, Y.; Strekalova, T.; Aquili, L.; Chang, R.C.; et al. Distribution and inter-regional relationship of amyloid-beta plaque deposition in a 5xFAD mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 964336. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-beta Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Yao, J.; Li, Z.; Zhou, Z.; Bao, A.; Wang, Z.; Wei, H.; He, H. Distinct regional vulnerability to Aβ and iron accumulation in post mortem AD brains. Alzheimer’s Dement. 2024, 20, 6984–6997. [Google Scholar] [CrossRef]
- Grothe, M.J.; Sepulcre, J.; Gonzalez-Escamilla, G.; Jelistratova, I.; Scholl, M.; Hansson, O.; Teipel, S.J.; Alzheimer’s Disease Neuroimaging Initiative. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 2018, 141, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Célestine, M.; Jacquier-Sarlin, M.; Borel, E.; Petit, F.; Perot, J.-B.; Hérard, A.-S.; Bousset, L.; Buisson, A.; Dhenain, M. Long term worsening of amyloid pathology, cerebral function, and cognition after a single inoculation of beta-amyloid seeds with Osaka mutation. Acta Neuropathol. Commun. 2023, 11, 66. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Kim, J.H.; Hasegawa, A.; Goto, R.; Sakai, K.; Ono, K.; Itoh, Y.; Yamada, M. Exogenous Aβ seeds induce Aβ depositions in the blood vessels rather than the brain parenchyma, independently of Aβ strain-specific information. Acta Neuropathol. Commun. 2021, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Riquelme, A.; Mao, A.; Barghash, M.M.; Lau, H.H.C.; Stuart, E.; Kovacs, G.G.; Nilsson, K.P.R.; Fraser, P.E.; Schmitt-Ulms, G.; Watts, J.C. Aβ43 aggregates exhibit enhanced prion-like seeding activity in mice. Acta Neuropathol. Commun. 2021, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Stonebarger, G.A.; Bimonte-Nelson, H.A.; Urbanski, H.F. The Rhesus Macaque as a Translational Model for Neurodegeneration and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 734173. [Google Scholar] [CrossRef]
- Purro, S.A.; Farmer, M.; Quarterman, E.; Ravey, J.; Thomas, D.X.; Noble, E.; Turnbull, C.; Linehan, J.; Nazari, T.; Brandner, S.; et al. Induction and characterisation of Aβ and tau pathology in AppNL-F/NL-F mice following inoculation with Alzheimer’s disease brain homogenate. BioRxiv 2024. preprint. [Google Scholar]
- Noda-Saita, K.; Terai, K.; Iwai, A.; Tsukamoto, M.; Shitaka, Y.; Kawabata, S.; Okada, M.; Yamaguchi, T. Exclusive association and simultaneous appearance of congophilic plaques and AT8-positive dystrophic neurites in Tg2576 mice suggest a mechanism of senile plaque formation and progression of neuritic dystrophy in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 435–442. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jaggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]

{kind=link}
| Host Animal | Aβ Seeds | Injection Site | Induced Pathological Features | Reference |
|---|---|---|---|---|
| APP23 mice | AβM1–42 fibrils AβM1–40 fibrils | Right cerebral hemisphere | Advanced Aβ plaques Glial activation Mild cognitive deficits | [82] |
| Aβ1–42 oligomers Aβ1–40 oligomers | Cortex or striatum | [79,91] | ||
| Aβ1–40 fibrils Aβ1–42 fibrils | Hippocampus | No detectable Aβ deposition | [77] | |
| Wistar rats | Aβ1–42 aggregates | Cerebroventricular | Memory impairment Neuronal death | [83] |
| Wistar rats (Rattus norvegicus) | Aβ1–42 oligomers | Lateral ventricle | No fibrillar amyloid deposits Tau phosphorylation | [85] |
| Cynomolgus macaques (Maccaca fascicularis) | Aβ1–42 oligomers | Lateral ventricle | No fibrillar amyloid deposits Increased Tau phosphorylation Increased neurofibrillary tangles Astrocyte and microglia activation Apparent synapse loss | [85] |
| Cynomolgus monkeys | Aβ1–42 oligomers | Brain parenchyma | Increased amyloid plaques Increased neurofibrillary tangles Profound neuroinflammation Degenerative neurons and synapses | [86] |
| Marmoset monkeys | Freshly dissolved Aβ1–40 or Aβ1–42 | Multiple sites * | No induced amyloid | [87] |
| Rhesus monkeys | Aβ1–42 oligomers | Cerebroventricular | Reduced spine density Microglia activation Induced neuroinflammation Increased CSF markers | [75] |
| Host Animal | Sources of Aβ Seeds | Injection Site | Induced Pathological Features | Reference |
|---|---|---|---|---|
| APP23 mice | 22–28 months APP23 mice | Hippocampus | Increase in Aβ deposition Microglia activation Astrocyte activation Dystrophic neurites | [5] |
| Hippocampus Entorhinal cortex Parietal cortex Striatum Olfactory bulb | Increase in Aβ deposition | [112,113] | ||
| 2 months APP23 mice 20–26 months APP23 mice 16 months APP/PS1 mice | Hippocampus | Increase in Aβ deposition Aβ-positive vessels Astrocyte activation Microglial activation Dystrophic neurites | [77] | |
| 20–27 months APP23 mice 20–27 months APP/PS1 mice | Hippocampus | Increase in Aβ deposition | [114] | |
| 18–30 months APP23 mice 18–30 months APP/PS1 mice | Peritoneal cavity | Increase in Aβ deposition | [115] | |
| 84 weeks APP23 mice | Hippocampus | Increase in Aβ deposition | [116] | |
| 25–27 months APP23 mice 20–22 months APP/PS1 mice | Hippocampus | Increase in Aβ deposition | [119] | |
| 2–28.6 months APP23 mice 1.2–22.1 months APP/PS1 mice | Hippocampus | Increase in Aβ deposition | [120] | |
| 24–26 months APP23 mice | Hippocampus | Increase in Aβ deposition | [123] | |
| APP/PS1 mice | 2 months APP23 mice 20–26 months APP23 mice 16 months APP/PS1 mice | Hippocampus | Increase in Aβ deposition | [77] |
| 15–18 months APP/PS1 mice | Sagittal midline Tail veins | Increase in Aβ deposition Vascular amyloid deposition | [122] | |
| 5×FAD mice | 51 weeks 5×FAD mice 14 weeks 5×FAD mice | Hippocampus | Increase in Aβ deposition | [116] |
| 21 months APP/PS1 mice | Hippocampus Entorhinal cortex | Increase in Aβ deposition Loss of NeuN | [117] | |
| 10 months 5×FAD mice | Hippocampus | Increase in Aβ deposition Astrocyte activation Microglial activation Demyelination | [118] | |
| Tg2576 mice | 18–20 months Tg2576 mice | Hippocampus Peritoneal cavity Thigh Eye | Increase in Aβ deposition Vascular amyloid deposition | [22] |
| APPNL-F/NL-F mice | 10 months TgCRND8 mice | Right parietal lobe | Increase in Aβ deposition Vascular amyloid deposition | [121] |
| R1.40 mice | 18–30 months APP23 mice 18–30 months APP/PS1 mice | Peritoneal cavity | Increase in Aβ deposition | [115] |
| 22–25 months APP23 mice | Hippocampus Overlying neocortex | Increase in Aβ deposition | [124] |
| Host Animal | Brain Donor of Aβ Seeds | Injection Site | Induced Pathological Features | Reference |
|---|---|---|---|---|
| APP23 mice | 74- and 85-year-old AD patients | Hippocampus | Increase in Aβ deposition | [77] |
| 69, 71, and 79-year-old AD patients | [145] | |||
| 62-, 81-, and 89-year-old AD patients | [146] | |||
| 61-, 64-, 62-, and 85-year-old AD patients | Right parietal lobe | Increase in Aβ deposition Increase in CAA Astrocyte activation Microglial activation | [93] | |
| Tg2576 mice | 84-year-old AD patient | Hippocampus Peritoneal cavity Thigh Eye | Increase in Aβ deposition Vascular amyloid deposition | [22] |
| 81-, 84-, and 91-year-old AD patients | Hippocampus | Increase in Aβ deposition Tau hyperphosphorylation Microglia activation | [125,147,148] | |
| APP/PS1 mice | 71–89-year-old AD patients | Hippocampus | Increase in Aβ deposition Induced tau pathology Neurofibrillary tangles Synaptic impairments Memory alteration Neuroinflammation | [149] |
| 76- and 83-year-old AD patients | Hippocampus | Increase in Aβ deposition Induced tau deposition | [151] | |
| Thy-Tau22 mice | 76- and 83-year-old AD patients | Hippocampus | Increase in Aβ deposition Induced tau deposition | [151] |
| Wistar rats | 77- to 87-year-old AD patients | Cerebroventricular | Decreased social memory Loss of volume in the LEC Neuroinflammation Synaptic reorganization | [21] |
| APP 21 rats | 78-year-old AD patients | Hippocampus | Increase in Aβ deposition | [152] |
| Mouse lemurs | 71–89-year-old AD patients | Posterior cingulate Cortex Corpus callosum | Increase in Aβ deposition Vascular amyloid deposition Neurofibrillary tangles Progressive cerebral atrophy Cognitive impairment | [150] |
| 76- and 83-year-old AD patients | Four different sites surrounding the parietal cortex | Increase in Aβ deposition Induced tau pathology Neuronal loss Cognitive impairments Modifications of neuronal activity | [151] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Song, S.; Liu, L.; Hong, W. In Vivo Seeding of Amyloid-β Protein and Implications in Modeling Alzheimer’s Disease Pathology. Biomolecules 2025, 15, 571. https://doi.org/10.3390/biom15040571
Liu Q, Song S, Liu L, Hong W. In Vivo Seeding of Amyloid-β Protein and Implications in Modeling Alzheimer’s Disease Pathology. Biomolecules. 2025; 15(4):571. https://doi.org/10.3390/biom15040571
Chicago/Turabian StyleLiu, Qianmin, Simin Song, Lu Liu, and Wei Hong. 2025. "In Vivo Seeding of Amyloid-β Protein and Implications in Modeling Alzheimer’s Disease Pathology" Biomolecules 15, no. 4: 571. https://doi.org/10.3390/biom15040571
APA StyleLiu, Q., Song, S., Liu, L., & Hong, W. (2025). In Vivo Seeding of Amyloid-β Protein and Implications in Modeling Alzheimer’s Disease Pathology. Biomolecules, 15(4), 571. https://doi.org/10.3390/biom15040571