
Peptide-Based Regulation of TNF-α-Mediated Cytotoxicity

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Peptide Synthesis and Analysis
2.1.2. Binding Affinity Experiments
2.1.3. In Vitro Experiments
2.2. Methods
2.2.1. Generation of Target Protein–Peptide Complexes
2.2.2. MD Simulations and Estimation of Relative Binding Free Energies Between Predicted Peptides and Target Proteins
2.2.3. Peptide Synthesis
2.2.4. Peptide Purification and Characterization
2.2.5. Microscale Thermophoresis
2.2.6. Cell-Based Assays
3. Results
3.1. Computational Prediction of Peptides and Dimeric Protein–Peptide Structures
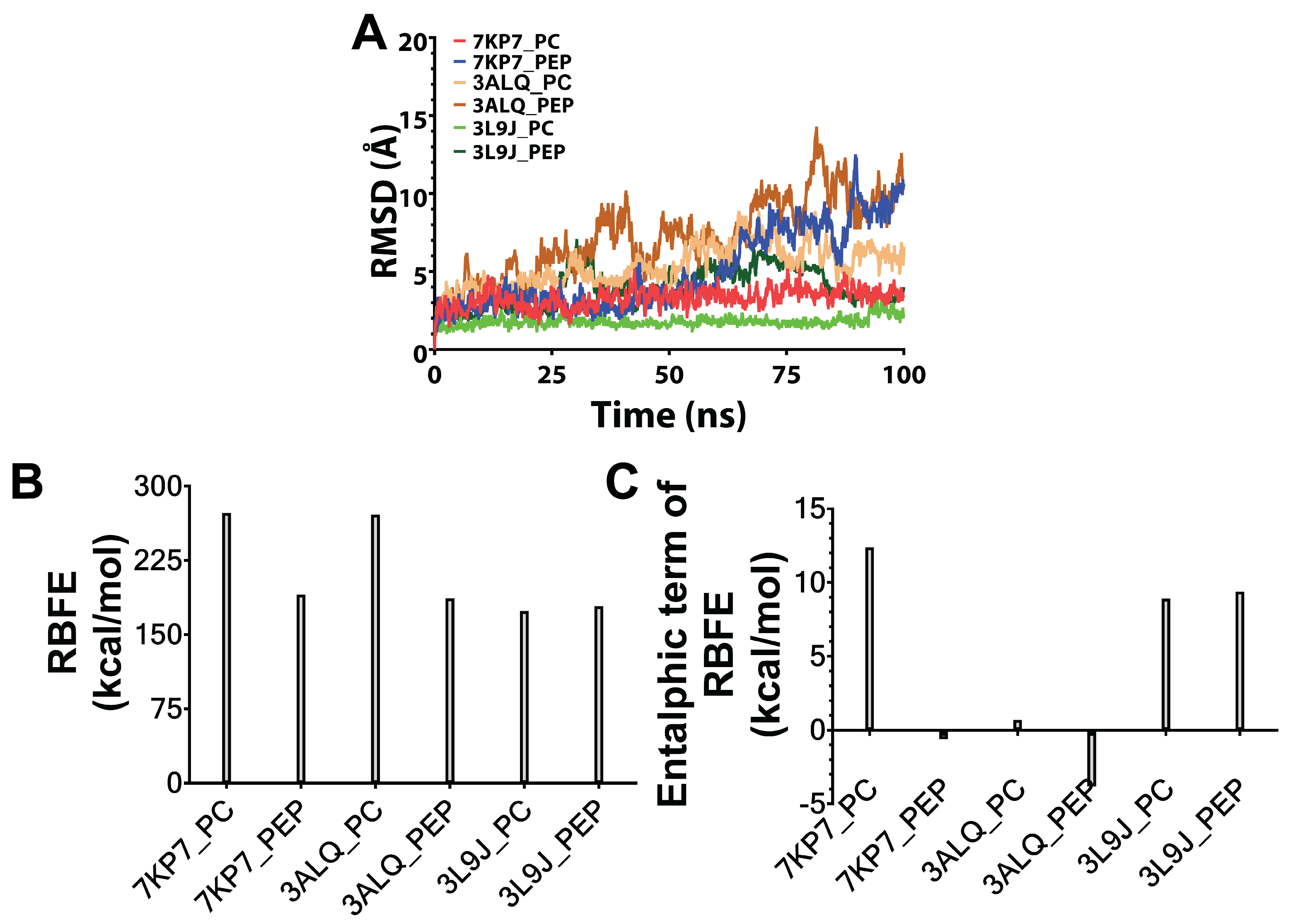
3.2. MD Simulations and Estimation of Relative Binding Free Energies Between Predicted Peptides and Target Proteins
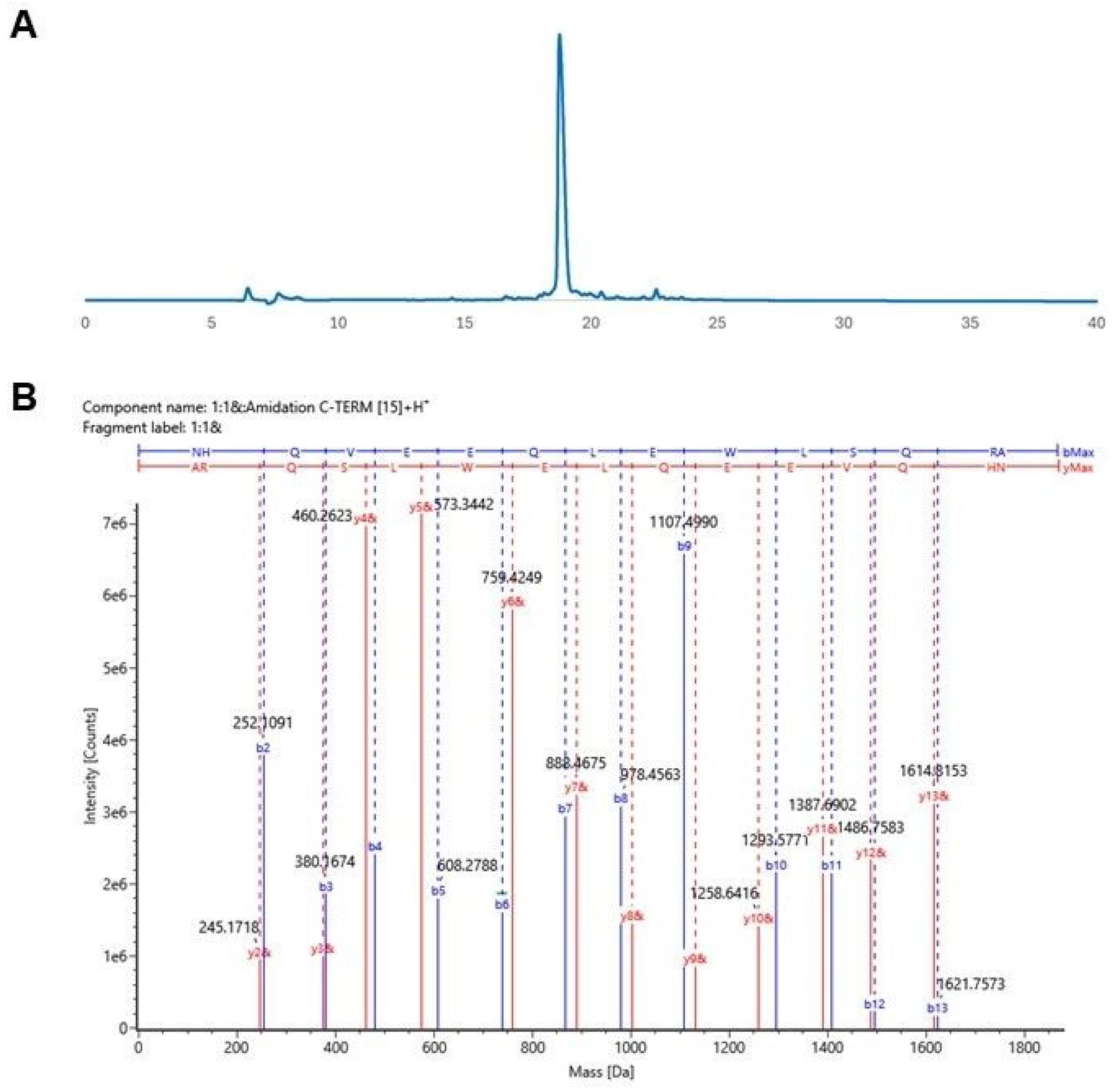
3.3. Synthesis and Characterization of Designed Peptides
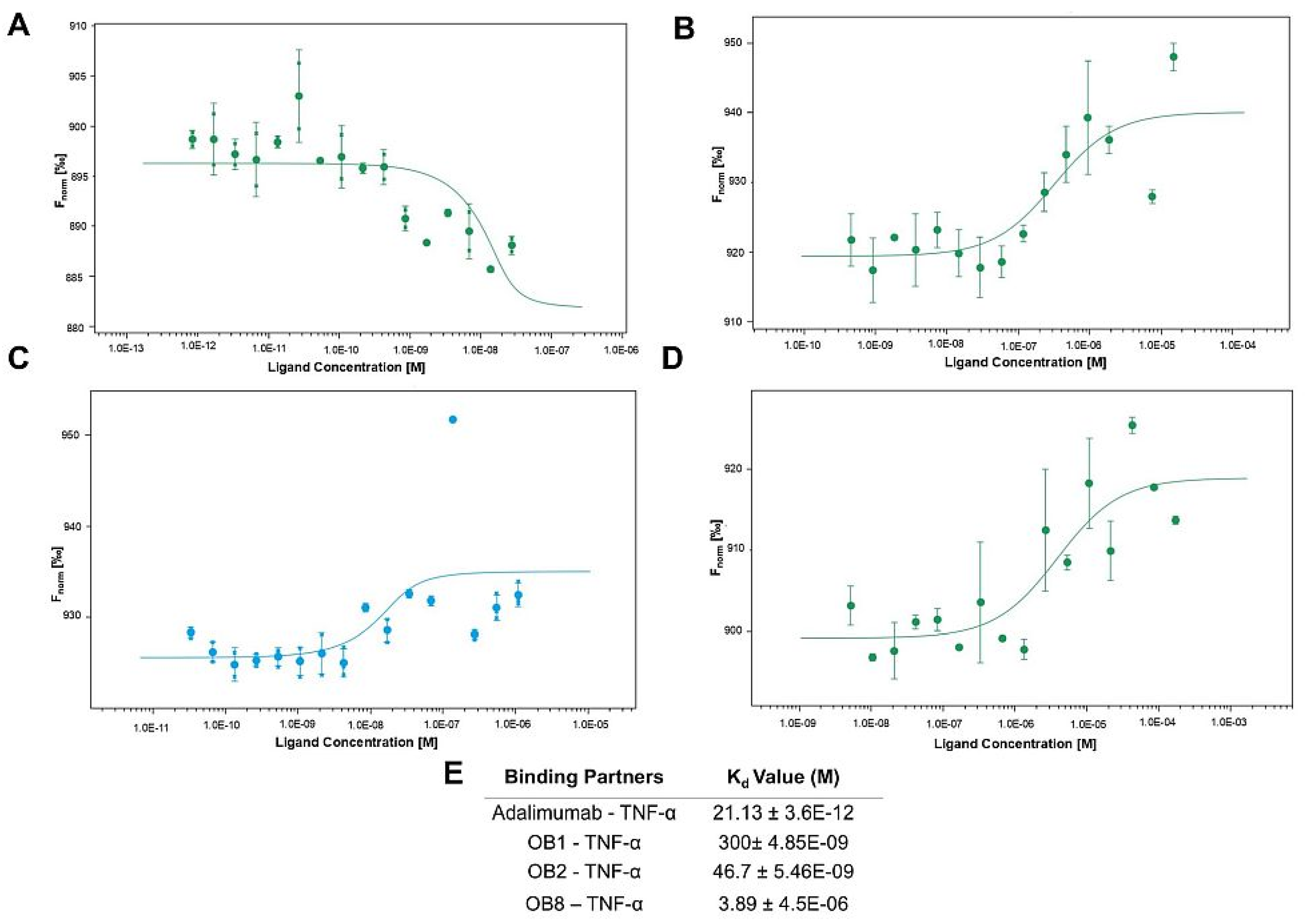
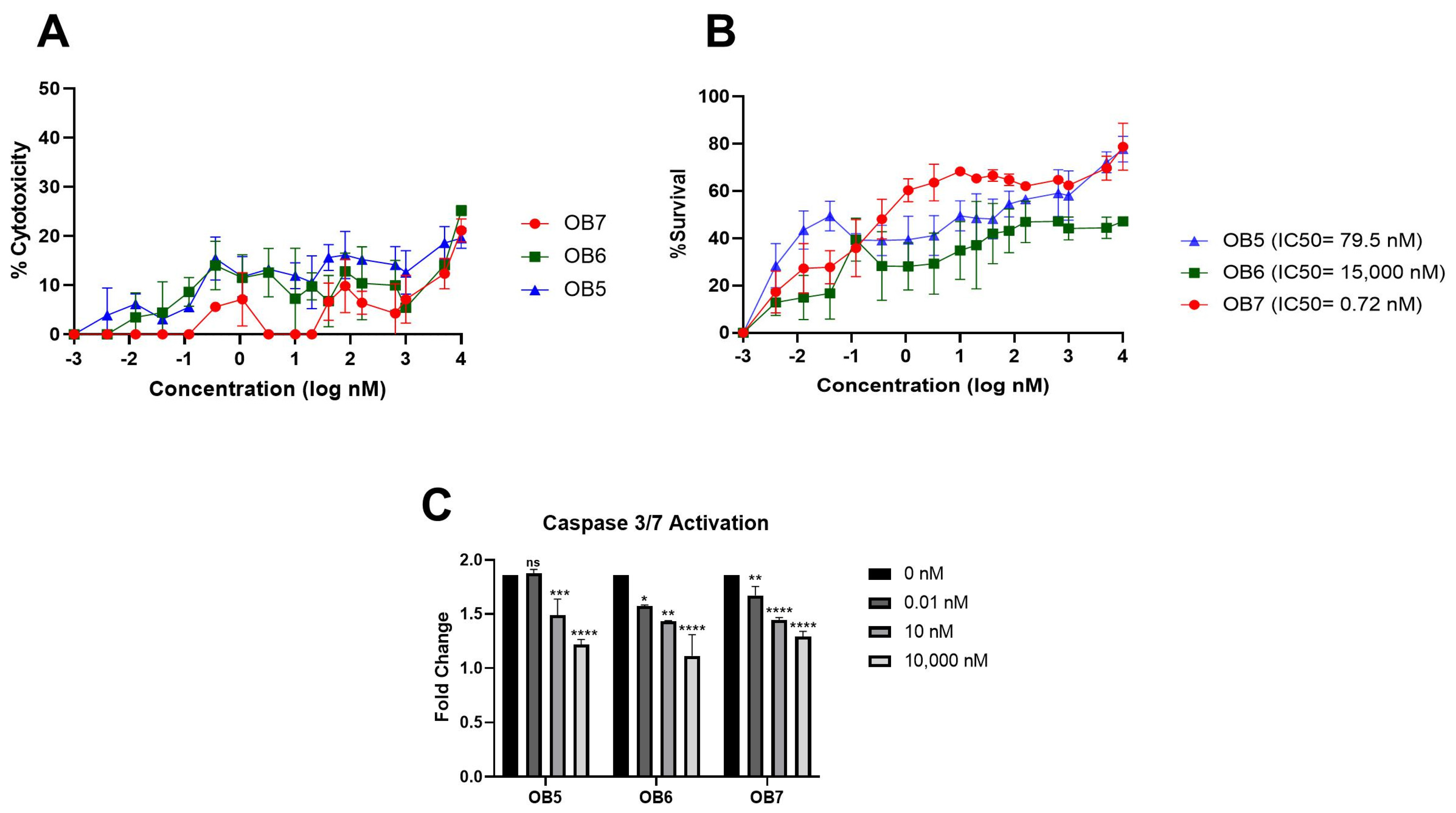
3.4. Binding Affinities of Designed Peptides for Their Target Molecules
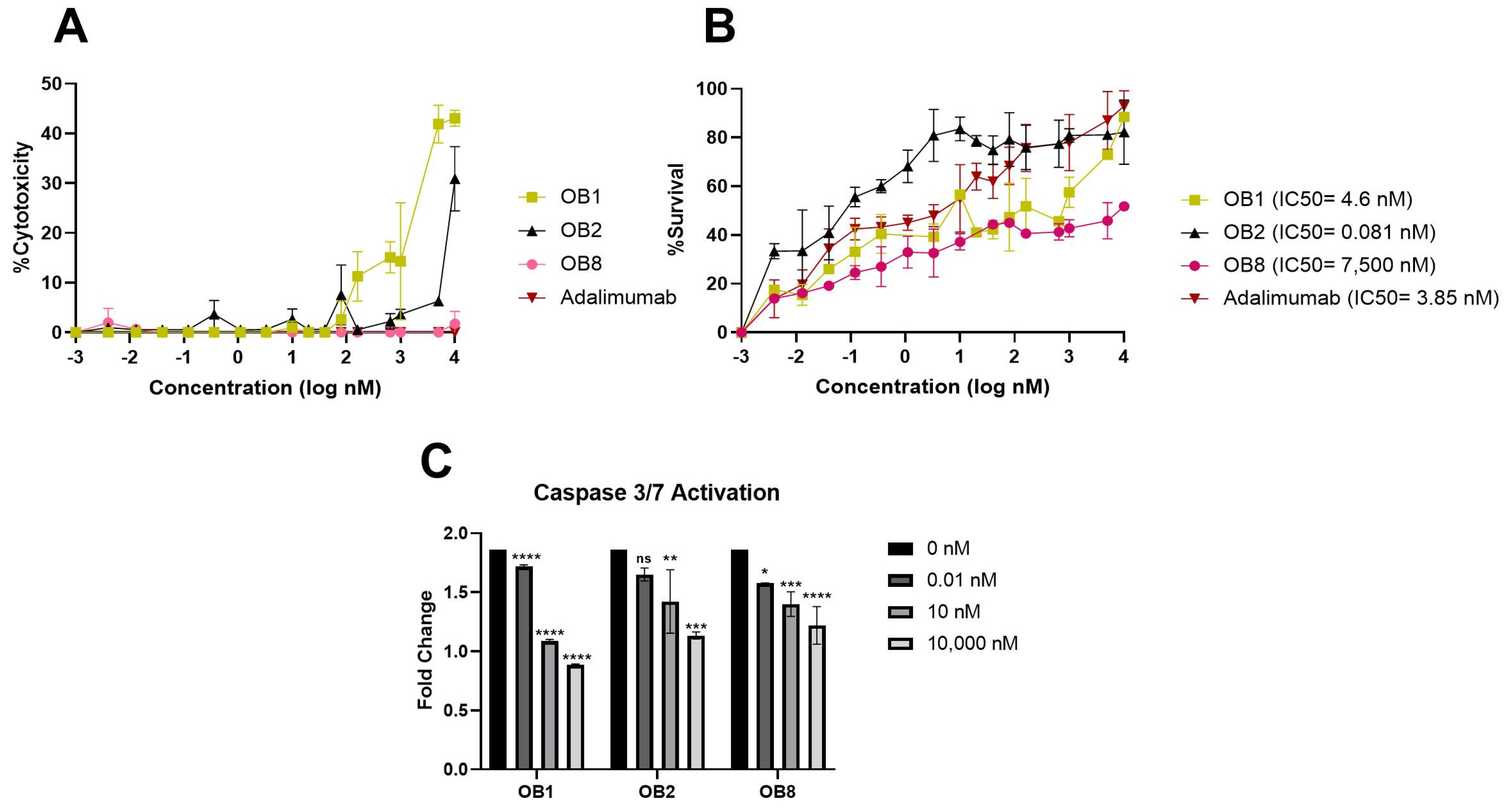
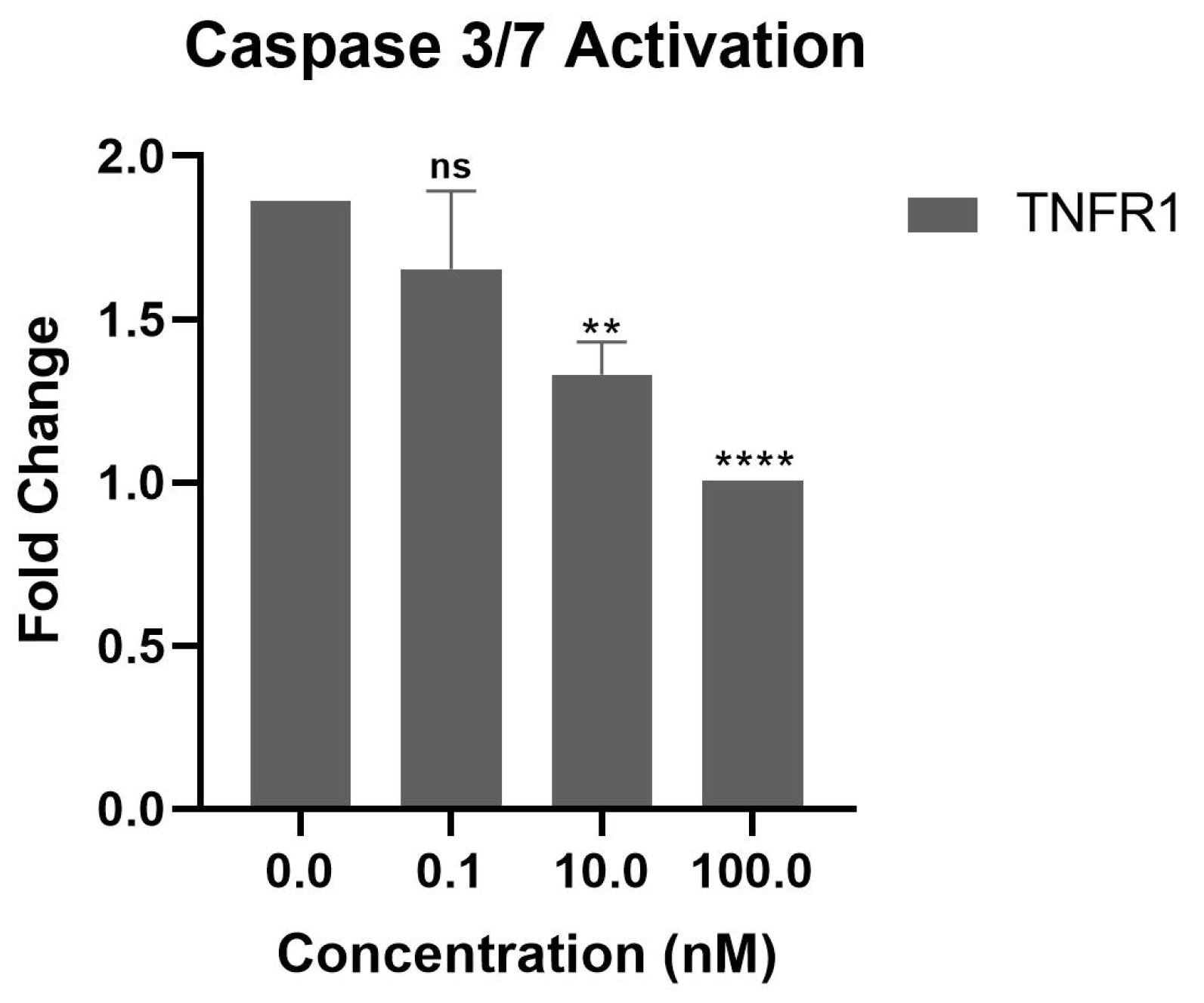
3.5. Inhibition of TNF-α-Mediated Cellular Cytotoxicity and TNFR Binding Using Synthesized Peptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TNF-α | tumor necrosis factor alpha |
| TNFR1 | TNF receptor 1 |
| TNFR2 | TNF receptor 2 |
| MST | microscale thermophoresis |
| TACE | TNF-α-converting enzyme |
| mAbs | monoclonal antibodies |
| PDB | Protein Data Bank |
| pLDDT | predicted local distance difference test |
| RMSD | root mean square displacement |
| SPPS | solid-phase peptide synthesis |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| Kd | dissociation equilibrium constant |
References
- Hehlgans, T.; Pfeffer, K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: Players, rules and the games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Griffiths, M.; Neal, J.; Gasque, P. Innate immunity and protective neuroinflammation: New emphasis on the role of neuroimmune regulatory proteins. Int. Rev. Neurobiol. 2007, 82, 29–55. [Google Scholar] [CrossRef]
- Wiens, G.D.; Glenney, G.W. Origin and evolution of TNF and TNF receptor superfamilies. Dev. Comp. Immunol. 2011, 35, 1324–1335. [Google Scholar] [CrossRef]
- van Horssen, R.; ten Hagen, T.L.M.; Eggermont, A.M.M. TNF-α in Cancer Treatment: Molecular Insights, Antitumor Effects, and Clinical Utility. Oncology 2006, 11, 397–408. [Google Scholar] [CrossRef]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, S.; Shen, S.; Fang, S.; Ye, Z.; Shi, Z.; Hong, A. A novel recombinant slow-release TNF α-derived peptide effectively inhibits tumor growth and angiogensis. Sci. Rep. 2015, 5, 13595. [Google Scholar] [CrossRef]
- Zia, K.; Ashraf, S.; Jabeen, A.; Saeed, M.; Nur-E-Alam, M.; Ahmed, S.; Al-Rehaily, A.J.; Ul-Haq, Z. Identification of potential TNF-α inhibitors: From in silico to in vitro studies. Sci. Rep. 2020, 10, 20974. [Google Scholar] [CrossRef] [PubMed]
- Kawalec, P.; Mikrut, A.; Wiśniewska, N.; Pilc, A. Tumor necrosis factor-α antibodies (infliximab, adalimumab and certolizumab) in Crohn’s disease: Systematic review and meta-analysis. Arch. Med. Sci. 2013, 5, 765. [Google Scholar] [CrossRef]
- Sharma, P.; Joshi, R.V.; Pritchard, R.; Xu, K.; Eicher, M.A. Therapeutic Antibodies in Medicine. Molecules 2023, 28, 6438. [Google Scholar] [CrossRef]
- D’Souza, A.; Meissner, B.L.; Tang, B.; McKenzie, R.S.; Piech, C.T. Effectiveness of Anti-Tumor Necrosis Factor Agents in the Treatment of Rheumatoid Arthritis: Observational Study. Am. Health Drug Benefits 2010, 3, 266. [Google Scholar]
- Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals 2021, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Malhotra, S.; Cameron, A.-I.; Gotham, D.; Burrone, E.; Gardner, P.J.; Loynachan, C.; Morin, S.; Scott, C.P.; Pérez-Casas, C. Novel approaches to enable equitable access to monoclonal antibodies in low- and middle-income countries. PLoS Glob. Public Health 2024, 4, e0003418. [Google Scholar] [CrossRef]
- Aboul-Ella, H.; Gohar, A.; Ali, A.A.; Ismail, L.M.; Mahmoud, A.E.E.-R.; Elkhatib, W.F. Monoclonal antibodies: From magic bullet to precision weapon. Mol. Biomed. 2024, 5, 47. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Zhao, X.; Ye, R.; Sun, L.; Wang, J.; Li, L.; Liang, H.; Wang, S.; Lu, Y. Discovery of an Anti-TNF-α 9-mer Peptide from a T7 Phage Display Library for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2023, 66, 6981–6993. [Google Scholar] [CrossRef]
- Highlights of Prescribing Iınformation of RINVOQ/RINVOQ LQ. Available online: https://www.rxabbvie.com/pdf/rinvoq_pi.pdf (accessed on 3 February 2025).
- Sedan, Y.; Marcu, O.; Lyskov, S.; Schueler-Furman, O. Peptiderive server: Derive peptide inhibitors from protein–protein interactions. Nucleic Acids Res. 2016, 44, W536–W541. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.L.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Madura, J.D. Solvation and Conformation of Methanol in Water. J. Am. Chem. Soc. 1983, 105, 1407–1413. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Martyna, G.J.; Hughes, A.; Tuckerman, M.E. Molecular dynamics algorithms for path integrals at constant pressure dynamics. J. Chem. Phys. 1999, 110, 3275–3290. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Crowley, M.F.; Williamson, M.J.; Walker, R.C. CHAMBER: Comprehensive support for CHARMM force fields within the AMBER software. Int. J. Quantum Chem. 2009, 109, 3767–3772. [Google Scholar] [CrossRef]
- Bulut, A.; Temur, B.Z.; Kirimli, C.E.; Gok, O.; Balcioglu, B.K.; Ozturk, H.U.; Uyar, N.Y.; Kanlidere, Z.; Kocagoz, T.; Can, O. A Novel Peptide-Based Detection of SARS-CoV-2 Antibodies. Biomimetics 2023, 8, 89. [Google Scholar] [CrossRef]
- Brunetti, J.; Lelli, B.; Scali, S.; Falciani, C.; Bracci, L.; Pini, A. A novel phage-library-selected peptide inhibits human TNF-α binding to its receptors. Molecules 2014, 19, 7255–7268. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H. Induction of apoptosis in TNF-Treated L929 cells in the presence of necrostatin-1. Int. J. Mol. Sci. 2016, 17, 1678. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, Z.; Wang, Y.; Ma, S.; Hu, Z.; Yang, P.; Chai, Y.; Xie, X. Discovery of Novel Ligands for TNF-# and TNF Receptor-1 through Structure-Based Virtual Screening and Biological Assay. J. Chem. Inf. Model. 2017, 57, 1101–1111. [Google Scholar] [CrossRef]
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.-I.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 2010, 3, ra83. [Google Scholar] [CrossRef]
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Kocagoz, T.; Temur, B.Z.; Unubol, N.; Elmas, M.A.; Kanlidere, Z.; Cilingir, S.; Acar, D.; Boskan, G.; Deveci, S.A.; Aybakan, E.; et al. Protease-Resistant, Broad-Spectrum Antimicrobial Peptides with High Antibacterial and Antifungal Activity. Life 2025, 15, 242. [Google Scholar] [CrossRef]
- Contreras, M.A.; Macaya, L.; Manrique, V.; Camacho, F.; González, A.; Montesinos, R.; Toledo, J.R.; Sánchez, O. A trivalent TNF-R2 as a new tumor necrosis factor alpha-blocking molecule. Proteins Struct. Funct. Bioinform. 2021, 89, 1557–1564. [Google Scholar] [CrossRef]
- Manrique-Suárez, V.; Macaya, L.; Contreras, M.A.; Parra, N.; Maura, R.; González, A.; Toledo, J.R.; Sánchez, O. Design and characterization of a novel dimeric blood–brain barrier penetrating TNFα inhibitor. Proteins Struct. Funct. Bioinform. 2021, 89, 1508–1521. [Google Scholar] [CrossRef]
- Ma, L.; Gong, H.; Zhu, H.; Ji, Q.; Su, P.; Liu, P.; Cao, S.; Yao, J.; Jiang, L.; Han, M.; et al. A novel small-molecule tumor necrosis factor α inhibitor attenuates inflammation in a hepatitis mouse model. J. Biol. Chem. 2014, 289, 12457–12466. [Google Scholar] [CrossRef]
- Abdulrahman, N.; Honda, T.J.; Ali, A.; Abdulrahman, N.; Vrinceanu, D.; Shishodia, S. Impacts of Indoor Dust Exposure on Human Colonic Cell Viability, Cytotoxicity and Apoptosis. Toxics 2023, 11, 633. [Google Scholar] [CrossRef]
- Napiórkowska, M.; Kumaravel, P.; Amboo Mahentheran, M.; Kiernozek-Kalińska, E.; Grosicka-Maciąg, E. New Derivatives of 1-(3-Methyl-1-Benzofuran-2-yl)Ethan-1-one: Synthesis and Preliminary Studies of Biological Activity. Int. J. Mol. Sci. 2024, 25, 1999. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.J.; Yang, J.; Lee, C.; Moon, Y.; Ahn, S.; Yang, J. Demonstration of functional similarity of a biosimilar adalimumab SB5 to Humira®. Biologicals 2019, 58, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Z.; Zhao, Y. Selective inhibition of Tumor necrosis factor receptor-1 (TNFR1) for the treatment of autoimmune diseases. Cytokine Growth Factor Rev. 2020, 55, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Moatti, A.; Cohen, J.L. The TNF-α/TNFR2 Pathway: Targeting a Brake to Release the Anti-tumor Immune Response. Front. Cell Dev. Biol. 2021, 9, 725473. [Google Scholar] [CrossRef]
- Lamers, C. Overcoming the Shortcomings of Peptide-Based Therapeutics. Future Drug Discov. 2022, 4, 75. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Molecular Weight (g/mol) | Binding Partner |
|---|---|---|---|
| OB1 | KGGGSGGGSHIHDDLLRYYGW | 2231.42 | rhTNF-α |
| OB2 | KGGGSGGGSGGGSRKEMGQV | 1748.90 | rhTNF-α |
| OB5 | NHQVEEQLEWLSQRA | 1866.02 | TNFR1 |
| OB6 | NPQAEGQLQWLNRRA | 1779.98 | TNFR2 |
| OB7 | VLLTHTISRIAVSYQTKVNLL | 2368.85 | TNFR1 |
| OB8 | KRWSRYFWVDMTGTR | 1988.31 | rhTNF-α |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temur, B.Z.; Timucin, A.C.; Atik, A.E.; Kocagoz, T.; Can, O. Peptide-Based Regulation of TNF-α-Mediated Cytotoxicity. Biomolecules 2025, 15, 559. https://doi.org/10.3390/biom15040559
Temur BZ, Timucin AC, Atik AE, Kocagoz T, Can O. Peptide-Based Regulation of TNF-α-Mediated Cytotoxicity. Biomolecules. 2025; 15(4):559. https://doi.org/10.3390/biom15040559
Chicago/Turabian StyleTemur, Betul Zehra, Ahmet Can Timucin, Ahmet Emin Atik, Tanil Kocagoz, and Ozge Can. 2025. "Peptide-Based Regulation of TNF-α-Mediated Cytotoxicity" Biomolecules 15, no. 4: 559. https://doi.org/10.3390/biom15040559
APA StyleTemur, B. Z., Timucin, A. C., Atik, A. E., Kocagoz, T., & Can, O. (2025). Peptide-Based Regulation of TNF-α-Mediated Cytotoxicity. Biomolecules, 15(4), 559. https://doi.org/10.3390/biom15040559